Article Text

Abstract
Background Mitochondrial or oxidative phosphorylation diseases are relatively frequent, multisystem disorders; in about 15% of cases they are caused by maternally inherited mitochondrial DNA (mtDNA) mutations. Because of the possible severity of the phenotype, the lack of effective treatment, and the high recurrence risk for offspring of carrier females, couples wish to prevent the transmission of these mtDNA disorders to their offspring. Prenatal diagnosis is problematic for several reasons, and concern the often poor correlation between mutation percentages and disease severity and the uncertainties about the representativeness of a fetal sample. A new option for preventing transmission of mtDNA disorders is preimplantation genetic diagnosis (PGD), which circumvents these problems by transferring an embryo below the threshold of clinical expression.
Methods We present the data on nine PGD cycles in four female carriers of mitochondrial diseases: three mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) (m.3243A>G), and one Leigh (m.8993T>G). Our threshold for transfer after PGD is 15% for the m.3243A>G mutation and 30% for the m.8993T>G mutation.
Results All four female carriers produced embryos eligible for transfer. The m.8993T>G mutation in oocytes/embryos showed more skewing than the m.3243A>G. In about 80% of the embryos the mutation load in the individual blastomeres was fairly constant (interblastomere differences <10%). However, in around 11% (in embryos with the m.3243A>G mutation only), the mutation load differed substantially (>15%) between blastomeres of a single embryo, mostly as a result of one outlier. The m.8993T>G carrier became pregnant and gave birth to a healthy son.
Conclusions PGD provides carriers of mtDNA mutations the opportunity to conceive healthy offspring.
- Clinical genetics
- Reproductive medicine
- Neuromuscular disease
- Diagnostics tests
- Molecular genetics
Statistics from Altmetric.com
Introduction
Mitochondrial or oxidative phosphorylation diseases are multisystem disorders, and in about 15% of cases they are caused by maternally inherited mitochondrial DNA (mtDNA) mutations.1 Carrier frequency for pathogenic mtDNA mutations in the normal population is high: one in 4002 to >1 in 200.3 Mitochondrial diseases can manifest with symptoms in many different organs, varying profoundly in severity and age of onset. Fatally affected newborns represent the severe end of the spectrum.
The majority of the severe pathogenic mtDNA mutations are heteroplasmic and characterised by a threshold effect, meaning that there are no symptoms unless the mutant load (proportion of mutant mtDNA) exceeds a certain level. This threshold varies both within tissues and between different mutations. In some mtDNA disorders, onset and severity of symptoms are clearly related to mutation load.4 ,5 Often, however, the phenotype and mutation load correlate poorly.6–8 MtDNA mutations are transmitted only from females to their offspring. The percentage heteroplasmy inherited by the fetus is affected by the ‘mitochondrial bottleneck’. During oogenesis the number of mtDNA molecules to be transmitted is reduced, and the resulting few mtDNAs become the founders for the offspring, resulting in considerable variation in mutant mtDNA among individual oocytes9 and subsequently among offspring. The ‘size’ of this bottleneck seems to depend on the type of mtDNA mutation10–12 and may even be individual-dependent for certain mutations.12
Mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS), mainly caused by the m.3243A>G mutation in the mitochondrial MT-TL1 gene, is a serious, common heteroplasmic mtDNA disorder.13 Age at onset, severity of symptoms, as well as organs involved are highly variable. Correlation between the level of mutant mtDNA in blood and clinical features is poor; however, mutation levels in muscle6 and urine14–17 seem to be of higher prognostic value. The level of mutant mtDNA in blood falls with time and, although rare, may become undetectable in women of fertile age.13 The frequency of affected offspring seems to be related to the level of mutant mtDNA in the mother's blood.18 Although segregation of the m.3243A>G mutation is thought to be largely determined by random genetic drift,11 ,12 the mutation percentage of the m.3243A>G mutation is reported to be higher in offspring than would be expected by random transmission only,19 ,20 which might be due to biases19 ,20 or sampling errors.21 On the other hand, a selection event against high mutant load might occur during meiosis and mitosis.22
A relatively common mtDNA mutation in children is the m.8993T>G mutation, leading to Leigh syndrome or NARP (neuropathy, ataxia, and retinitis pigmentosa), depending on mutant load,23 which correlates rather well with the phenotype.4 Also the risk of affected offspring can quite accurately be predicted by the mother's mutation load.4 The transmission of the m.8993T>G mutation is generally skewed with an overrepresentation of oocytes with 0% and 100% mutation load.4 ,10 ,23 ,24 As for the m.3243A>G mutation, the possibility of preferential mutant genome transmission has also been proposed for the m.8993T>G.19 ,20
Because of the high frequency of mitochondrial disorders, the possible severity of the phenotype, the lack of effective treatment, and the high recurrence risk for offspring of carrier females, couples may aim to prevent transmission. A key problem in prenatal diagnostics (PND) for mitochondrial disorders is the unreliable correlation between the mutant load and the disease severity, making it difficult to predict the clinical severity of the disease in the child and the likelihood of a couple having affected offspring.13 Furthermore, data on the representativeness of chorionic villi or amniocytes for the mutant load in the different fetal tissues are contradictory.4 ,7 ,12 ,25–28 Mutant load distribution between different fetal tissues and segregation throughout the embryo–fetal development does not seem to be a restriction.4 ,12 ,23–26 29–38
A fairly new option for preventing transmission of mtDNA diseases is preimplantation genetic diagnosis (PGD).12 ,24 ,39–41 In PGD, embryos obtained after in vitro fertilisation (IVF) are analysed, and only those with amounts of mutant mtDNA below the predicted threshold of (severe) expression are transferred to the uterus. Uncertainties concerning this threshold make it difficult to develop PGD for all mtDNA mutations, although a recent meta-analysis provided some guidelines.41 Our threshold for the m.3243A>G mutation (MELAS) is 15% and for the m.8993T>G (Leigh) 30%, based on data of muscle mutation load and clinical manifestations,4 ,6 assuming that the muscle mutation load correlates with the embryonic mutation load, and with a safety margin to correct for potential errors in determining heteroplasmy levels.
We present data on nine PGD cycles in four female carriers of mitochondrial disease; three women with the MELAS m.3243A>G mutation and one with the Leigh m.8993T>G mutation. Worldwide, very few centres perform PGD for mitochondrial diseases,12 ,24 ,40 and our data will help in understanding the dynamics of mtDNA mutations, evaluating the suitability of PGD for mtDNA disorders, and optimising guidelines regarding the PGD procedures.
Methods
The study was approved by the local ethical committee.
Patients
Three couples with the m.3243A>G mutation and one with the m.8993T>G mutation were counselled for PGD. Couples received verbal and written information on IVF and intracytoplasmic sperm injection (ICSI), single cell procedures, success rates of the IVF/PGD treatment, risks of misdiagnosis, and the worldwide limited experience of PGD for mtDNA disorders. Prenatal diagnosis to confirm the PGD diagnosis was offered. Informed consent was given by both partners before treatment, including consent for re-analysis of non-transferred and non-frozen embryos. The threshold level for transfer, being 15% for the m.3243A>G and 30% for the m.8993T>G mutation, was discussed with the couples. The couples could not opt for transfer of embryos above that threshold, for example, in case none of the embryos would have mutation loads below the threshold. The Leigh couple requested transferral of only embryos with 0% mutation load, but after counselling a threshold level for transfer of 5% was agreed upon as no severe symptoms will occur at that level. The female carriers underwent a clinical examination to determine contraindications for the IVF procedure. Mutation loads in blood, hair, urine and/or muscle of the carrier females were established.
IVF/ICSI/PGD procedure
Oocytes were retrieved under ultrasound guidance, following controlled ovarian stimulation.42 After 5 h of maturation, MII oocytes were fertilised with ICSI43 followed by embryo culturing.44 At day 3 post-fertilisation, blastomeres were biopsied, one blastomere from 4–6-cell stage embryos and two blastomeres from 7-cell stage and beyond. Biopsies were performed in Ca2+ and Mg2+ free medium with a micromanipulator (Narishige, ONO-121, Paes Nederland, Zoeterwoude, The Netherlands) mounted on an inverted microscope (Olympus, IX-70, Paes Nederland). Biopsied blastomeres were washed three times in phosphate buffered saline solution with 1% polyvinylpyrrolidone molecular weight 360 000 (Sigma-Aldrich Chemie, Zwijndrecht, the Netherlands) and 0.1 mg/ml phenol red (Sigma), and transferred to a 0.2 ml PCR reaction tube. Blank samples were taken from the last washing droplet. After PGD analysis, one (preferably) or two embryos with a mutation load below the threshold were selected for embryo transfer at day 4 after fertilisation. Selection was, besides mutation load, based on embryo score criteria.44
PCR and single cell quantification assay
PCR procedure m.3243A>G and 8993T>G mutations
The alkaline lysis buffer was decontaminated from exogen DNA by 1 h ultraviolet C (UV-C) irradiation using a UV-C lamp type TUV 30W/G30T8 long life (Philips). The PCR mix without the DNA-polymerase was decontaminated by MnlI restriction enzyme (5 U/μl; Biolabs) incubation for 2 h at 37°C followed by 30 min inactivation at 65°C. Blank samples were included in every PCR series to monitor DNA contamination. Cells were lysed by adding alkaline lysis buffer (50 mM dithiothreitol (DTT) (Pharmacia Biotech)/200 mM NaOH) and 10 min incubation at 65°C. After cell lysis, PCR was performed on the GeneAmp PCR System 9700 (Perkin-Elmer Applied Biosystems) in a total volume of 50 μl, containing 1 X PCR buffer (Invitrogen) and 1 U of Taq DNA polymerase (Invitrogen), 2 mM MgCl2 (Invitrogen), and 0.1 mM dNTP (Pharmacia). To neutralise the alkaline lysis buffer Tricine (20 mM pH 4.95 (Sigma)) was used. Unlabelled forward and reverse primer sets were used in the first round and varied from 3–15 pmol primer per sample (table 1).
Primers used for PCR amplification of the m.3243A>G and m.8993T>G mutations and the m.8993T>G digestion control amplicon
First round PCR started with 5 min denaturation at 94°C followed by 38 cycles of 1 min denaturation at 92°C, 45 s annealing at 53°C, and 45 s elongation at 72°C, followed by a final elongation step of 7 min at 72°C. 15 µl of first round amplification product was adjusted to 50 µl with second round PCR mix, containing 1 X PCR buffer (Invitrogen) 1 U of DNA polymerase, 2 mM MgCl2 (Invitrogen) and fluorescently labelled primer (table 1). One final amplification cycle was performed of 5 min denaturation at 94°C, 1 min annealing at 53°C, and 7 min elongation at 72°C.
The labelled second round PCR product (15 µl) was digested in a total volume of 50 µl containing 10 U with HaeIII (10 U/µl; Biolabs) for the m.3243A>G mutation and HpaII (50 U/µl; Biolabs) for the m.8993T>G mutation. The m.3243A>G-amplicon contains an additional HaeIII restriction site as an internal control for restriction enzyme digestion completion. The digestion sample of the m.8993T>G is spiked with a labelled amplicon generated by the HpaIINARP (6-FAM) and HpaIINARP-R primers containing an HpaII site as control. After digestion samples were purified using a QIAquick PCR purification kit (Qiagen). Samples were analysed by capillary electrophoresis on an ABI Prism 3730 Genetic Analyser followed by GeneScan analysis.
Single cell (semi)quantification and statistical analysis
To calculate the mutation load the area of the mutation peak was divided by the sum of the peak area of the wild type and mutation peak.12 ,45 Five reference samples with a mutation load around the threshold value were included in every series for validation. The reference amplicons were diluted to a single blastomere equivalent amount, approximately 12 500–75 000 mt DNA copies.46 A total of 50 of these reference samples were amplified in 10 separate PCR runs during protocol setup and the data were used to construct a Shewart chart47 (figure 1A,B, see online supplementary data), which showed the mean, the mean±1 SD, the mean±2 SD, and the mean±3 SD. Four of five control observations must fall within the mean±2 SD to approve the PGD PCR analysis. Data were rejected if more then one control sample exceeded the mean±3 SD limit. Due to the linearity of amplification for the different percentages of mutation loads12 ,45 (figure 2A,B, see online supplementary data) only control samples around the threshold mutation load were used for constructing a Shewart chart.

Pedigrees with family members showing clinical features and mutant loads in blood (Bl), hair follicles (H), muscle (M), urine (U), and/or fibroblasts (Fibr).

Preimplantation genetic diagnosis cycles of the respective carrier females. Each cluster of bars represents an embryo with its tested blastomeres. The red dotted line represents the threshold level for transfer. For the Leigh carrier, the embryos in which the mutation was not detected are depicted as X. For these embryos the numbers of analysed blastomeres are not visible in the figure, but are available in online supplementary data. ET, embryo transfer.
Analysis of oocytes and remaining embryos
Surplus embryos not suitable for transfer or cryopreservation were collected on day 4. The zonae pellucidae were removed by incubation in 1 U/µl pronase (Sigma, Zwijndrecht, The Netherlands). In embryos that could not be dissected in separate blastomeres due to strong intercellular interactions once in the morula stage, it was generally possible to identify the number of blastomeres clustered and the mean mutation load was calculated. Remaining fertilised oocytes, zygotes, embryos or single blastomeres were washed three times in washing buffer, transferred to a 0.2 ml PCR reaction tube, and stored at −20°C until PCR.
Results
Patients and PGD cycles
The four women were aged 36, 30, 28, and 30 years at the start of the first cycle.
Pedigrees and mutation loads are given in figure 1 below and table S1 (see online supplementary data). The four women did not show any symptoms or abnormalities upon clinical examination. The only probable exception is hearing loss of high frequencies in the first m.3243A>G carrier. The first m.3243A>G couple had been infertile for many years. The female carrier had a severely affected mother. The second and third m.3243A>G carriers had severely affected brothers, who died at young adult age. The Leigh/NARP couple lost their first child affected by Leigh syndrome in the first year of his life. Nine PGD cycles were performed in total: three, two, and two cycles for each m.3243A>G carrier, respectively, and two for the m.8993T>G carrier. The nine cycles resulted in seven transferred embryos (six transfer procedures) (figure 2); two embryos in the first m.3243A>G carrier, one in each of the other m.3243A>G carriers, and three in the m.8993T>G carrier, eventually leading to one pregnancy of the latter. PND was refused. Pregnancy follow-up was performed by a gynaecologist. A healthy son was born. Cord blood analysis confirmed the initial PGD outcome; no m.8993T>G mutation was demonstrated.
Mutation analysis in embryos, oocytes, and zygotes
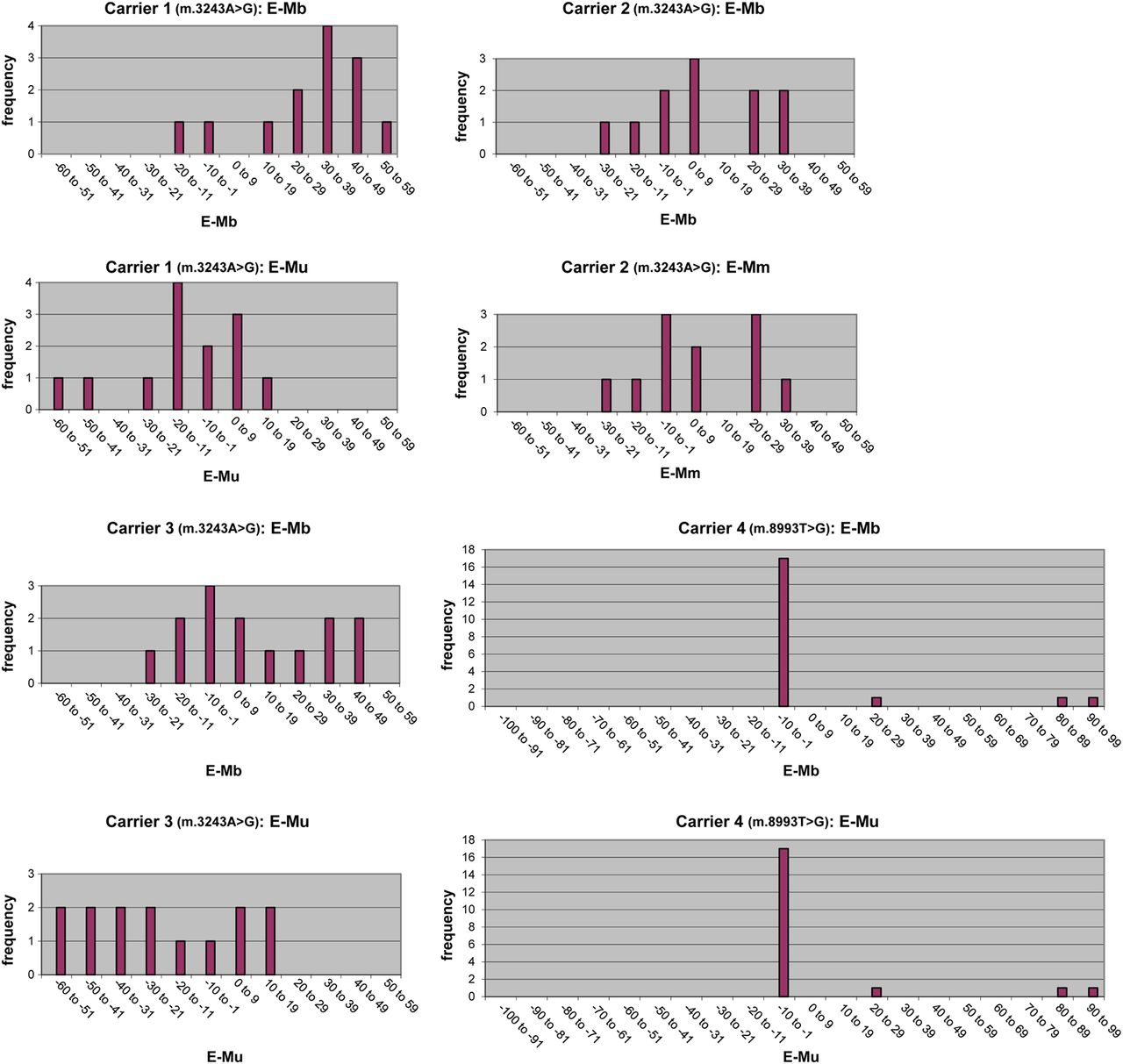
A total of 58 embryos, nine oocytes, and 20 zygotes (of which nine were degenerated) could be analysed (figure 2) (table S1 supplementary data). The m.3243A>G mutation was detected in four of four oocytes and in 16 of 18 zygotes (see online table S1 supplementary data) and in all 38 embryos (100%, figure 2) from the three carriers. Oocyte mutant load was 37–67% in the first MELAS carrier (three oocytes analysed) and 65% in the second carrier (one oocyte analysed). Zygote ranges were 0–66% (11 zygotes analysed), 15–42% (four zygotes analysed), and 0–66% (three zygotes analysed), and the average mutation load in the 38 remaining m.3243A>G embryos ranged from 1–65%, 2–58%, and 5–70% for the three women, respectively (see online table S1 supplementary data). The m.8993T>G mutation was only present in one of five oocytes (mutation load 16%, see online table S1 supplementary data) and in three of 20 embryos (average mutation load 30–100%, figure 2). The histograms concerning the difference in average mutation load between offspring (preimplantation embryos) and the carrier females are shown in figure 3.
{kind=link}
{kind=link}
{kind=link}
Histograms of the difference in average mutant load between the preimplantation embryos (E) and the corresponding carrier females (M). Mb, mutation load in blood; Mm, mutation load in muscle; Mu, mutation load in urine.
Interblastomere differences of mutation load within embryos
Interblastomere mutation load differed <10% in most embryos (43 out of 53; 81%), but in six of 53 tested embryos (11%) it differed more than 15%. These were embryos 2.1 (ie, cycle 2, embryo 1), 3.4, and 3.5 of the first m.3243A>G carrier and embryos 1.4, 2.1, and 2.2 of the second (figure 2), displaying a range of 60–82%, 50–65%, 35–54%, 21–38%, 54–77%, and 5–30%, respectively. So, approximately 18% of the m.3243A>G embryos showed an interblastomere variation of >15%. In four additional embryos with the m.3243A>G mutation, we found interblastomere mutation load differences of >10%. In almost all cases, there was only one outlier responsible for the large range. The only exception was embryo 3.5 of carrier female 1 (figure 2), in which the large range was due to differences between several blastomeres rather than one outlier.
Discussion
Transmission rates 3243A>G and 8993T>G mutation
The m.3243A>G mutation was detected in the great majority of oocytes, zygotes and embryos examined, which corroborates earlier reports in which the m.3243A>G was detected in 90% (74/82) of the primary oocytes of an adult carrier11 and in 84% (32/38) of day 3 stage embryos.12 In contrast, the m.8993T>G mutation was detected in only a minority of oocytes and embryos examined. Our results differ from Blok et al10 who analysed seven oocytes from a 8993T>G mutation carrier with 50% mutation load in blood and found only one oocyte lacking the mutation. This is likely due to the difference in maternal mutation load, which is only 4% in the blood in our patient, supporting the observation that the individual recurrence risk, and thus the transmission rate for this mutation, can be quite accurately predicted.4
Division of mutation load among oocytes and embryos: segregation patterns
The large variation in mutant load among oocytes and embryos of the m.3243A>G carriers that we observed is in line with the percentages of 0–77% found by Monnot et al.12 These data, approximating a Gaussian curve (data not shown, derivable from figure 3), support the proposal that the level of mutant mtDNA in human primary oocytes and embryos is largely determined by random genetic drift.11 ,12 Embryonic mutant load that appeared to be higher compared to the mother's mutation load in blood (first and third m.3243A>G carrier, figure 3), appeared on the contrary lower when compared with the mother's mutation load in urine (figure 3). Our data suggest that the reported preferential transmission of mutant genomes may, at least for the m.3243A>G mutation, reflect a positive bias due to differences in mutation load between blood and urine/muscle of carrier females. Based on the longitudinal decrease in m.3243A>G mutant load in blood,48–50 the difference in mutant load between offspring and their corresponding mothers could be overestimated.19 ,20 However, in only a few of the published pedigrees were muscle data available, and no urine data at all. Furthermore, our results support the hypothesis that a selection event against very high m.3243A>G mutant load in oocytes and subsequent embryos might occur during meiosis.12 ,22
A predominantly skewed segregation of the m.8993 mutations during the preimplantation/prenatal period has been reported.10 ,24 The majority of our embryos indeed had no mutation, which was expected from the low maternal mutation load. However, two embryos had an intermediate mutation load (88% and 30%), which has been reported previously.40 These findings seem to contradict the hypothesis of highly skewed segregation, but are in accordance with intermediate values of the m.8993A>G mutant load observed after birth. Individual factors influencing a given mutation's bottleneck behaviour might contribute to this incomplete skewing, as has been hypothesised before for the m.3243A>G mutation.22
Relation to mutation load in the mother: recurrence risk
The frequency of affected offspring is related to the level of mutant mtDNA in the mother's blood for the m.3243A>G mutation.18 The risk of clinically affected offspring is in our data higher for the first m.3243A>G carrier, with 13% in blood, than for the second, with 25%. This was unexpected based on risk estimates in previous reports.12 ,18 These data emphasise that, although in general a higher mutant load in the mother provides a higher risk of affected offspring,18 the recurrence risk for an individual m.3243A>G carrier remains very difficult to predict. Furthermore, our data underline the conclusion that mothers carrying the m.3243A>G mutation have a high frequency of affected offspring whatever the level of mutant mtDNA in their blood.18 Possibly a better prediction can be made based on the mother's mutation load in muscle/urine, but more data are necessary to determine this more accurately.
For the m.8993T>G/C mutations the recurrence risk, defined as the probability of a severe outcome, is related to the mutation load of the carrier mother4 and would for our carrier be approximately 13%, but with a wide range. This is grossly reflected by our data; the mutation was present in 15% (3/20) of m.8993T>G embryos, in two of them above the threshold of clinical expression.
Interblastomere differences in mutation load
Mutation load was comparable between blastomeres in the majority of the embryos. In the m.3243A>G subgroup, 18% of affected embryos showed an interblastomere variation of >15%. One outlier was responsible for the large range in almost all cases. Blastomeres of mouse embryos have been reported to contain very similar levels of variant mtDNA.37 ,51 ,52 Steffan et al24 found a small (≤8%) intercellular variation of mitochondrial heteroplasmy among 12 of 15 human embryos tested for an mtDNA polymorphism. In the remaining three embryos, all originating from the same individual, the intercellular variation was 16–19%. This, combined with our data in which large intercellular variation occurred in only two of three m.3243A>G carriers, suggests that genetic factors could be involved in mitochondrial segregation during embryogenesis.24 Monnot et al12 found an equal distribution of mutant DNA among various blastomeres of a carrier embryo until day 5, with only occasional variability up to 15% in mutant load between single cell measurements and the whole embryo; this was assumed to result from technical artefacts, or, less likely, physiological distortion in m.3243A>G segregation from day 3 to day 5. However, in one embryo we found a large variation at initial biopsy instead of at re-analysis, excluding segregation from day 3 to day 5 as an explanation. In our series, interblastomere differences were not only larger, they also occurred more often than previously reported,12 although Monnot et al did not perform single blastomere analysis in all embryos. Interestingly, a large interblastomere variation of 24% in an m.3243A>G embryo has been reported by another group.53
Differences in mutation load between blastomeres would not be a problem if all values remained above or below the threshold level. However, in one embryo, we found one outlier which would make the embryo eligible for transfer, whereas in fact it was not. Applying a threshold of 30%12 would have caused the same situation in two other embryos (1.4 and 2.3, see online table S2 supplementary data). A false-negative classification can be minimised by analysing two blastomeres, instead of one as proposed before,12 although the removal of two cells has been reported to influence the rate of live birth delivery.54 This illustrates the difficult balance between a safe and correct diagnosis on the one hand, and optimising the chance of pregnancy on the other.
Conclusion
Our data from nine PGD cycles in four female carriers of mitochondrial diseases—three MELAS (m.3243A>G) and one Leigh case (m.8993T>G)—show that PGD for mtDNA disorders is useful as all carriers produced oocytes below the threshold. Blastomere mutation load appears to be representative for the whole embryo in the majority of cases. More data are necessary to understand further the segregation of mtDNA mutations throughout human embryo-fetal development, and to optimise guidelines regarding the PGD procedures. Counselling of couples who are at high risk of having offspring with an mtDNA disorder is still a challenge. However, PGD provides those couples with the opportunity to conceive healthy offspring.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figure 1
- Data supplement 2 - Online figure 2
- Data supplement 3 - Online table
Footnotes
-
Contributors SCEHS: collected and analysed the data of the performed PGD cycles, studied the literature, wrote the article except part of the Methods section (main authorship). JCFMD: coordinated and authorised the genetic laboratory work concerning the PGD cycles, wrote (main authorship) the technical parts of the Methods section of the article, carefully reviewed the article (main authorship). MD and SS: performed genetic laboratory work, reviewed the article. FHJvT: performed practical work on the MELAS mutation. EC: involved in the IVF procedures, reviewed the article. RJTvG: involved in the IVF procedures, reviewed the article. IFMdC: referred patients for PGD, reviewed the article. JPMG: head of department of Clinical Genetics and PGD Maastricht, reviewed the article. CEMdD-S: medical coordinator PGD, counselled patients for PGD, supervised writing of the article. HJMS: head of department Clinical Genomics, authorised the genetic laboratory work concerning the PGD cycles, supervised writing of the article. CEMdD-S and HJMS are shared last author. All authors approved the final version.
-
Competing interest None.
-
Patient consent Obtained.
-
Ethics approval Local ethics committee, Maastricht University Medical Centre.
-
Provenance and peer review Not commissioned; externally peer reviewed.