Article Text

Statistics from Altmetric.com
Research into the molecular and genetic basis of disease is continually expanding and improving the prospects for rational treatments. Of these, gene therapy (here defined as the introduction of genetic material into human cells) may ultimately offer the greatest scope.1 This article provides a perspective on the potential for gene therapy for treating inherited retinal degenerations, along with an outline of progress and problems encountered to date. Of the inherited forms of retinal degeneration, retinitis pigmentosa (RP) is the best characterised (see Bird2 for review). Seven different non-syndromic RP genes have been identified to date, five of which3-7 are expressed exclusively in photoreceptor cells. However, photoreceptors and retinal pigment epithelial (RPE) cells are in close proximity and are interdependent. Defects in genes expressed in the RPE may also result in retinal degeneration. Delivery to these tissues of either normal copies of the defective genes or genes which enhance cell survival may arrest the degenerative process and thus preserve vision.
THE EYE AS TARGET ORGAN FOR GENE DELIVERY
The eye has a number of advantages as a target organ for gene delivery. It is easily accessible and the tissues may be examined in vivo by ophthalmoscopy. In addition, there are blood-retinal and blood-aqueous barriers which may concentrate vectors in the target area and reduce their spread out of the eye. The eye may be used for testing gene delivery to a wide range of tissues since it contains endothelium (cornea), epithelium (cornea, ciliary body, iris), muscle (ciliary body), and neurons (retina). It may also serve as a valuable model system to test gene therapy strategies for the brain, whose neurons are more difficult to target than those in the neuroretina.
SCOPE OF GENE THERAPY TO TREAT OCULAR DISORDERS
It is probable that gene therapy for certain ocular diseases will be realised sooner than others. Gene therapy strategies for cancers (for example, uveal melanoma), infections (for example, cytomegalovirus retinitis (CMV) retinitis), inflammation (for example, uveitis, proliferative vitreoretinopathy), and as an adjuvant to surgery (for example, to reduce scarring) probably only require short term gene expression which is not critically dependent upon precise regulation. However, gene therapy for inherited retinal degenerations, where there is a requirement for long term gene expression with appropriate regulation, will present considerable difficulties. Nevertheless, there are many monogenic disorders involving retinal degeneration and they are probably the most appropriate ocular disease candidates for gene therapy since they are at present untreatable—other genetic disorders such as familial cataracts or corneal dystrophies may be reasonably adequately treated by surgery/transplant. Although three main treatment strategies for retinal degenerations are currently under investigation in animals—replacement of photoreceptors8-12 or RPE13-15 by transplantation, rescue by intraocular injection of growth factors,1617 and restoration of function by gene delivery to the appropriate cells18 —the third strategy, gene therapy, potentially offers the most precise treatment. It aims in recessive disease to introduce a functional copy of the defective gene or in dominant diseases (other than in cases of haploinsufficiency) to specifically inactivate messages from the defective gene. Although there are many problems to be overcome before gene therapy becomes a viable treatment, the problems are fairly well defined, and each may be investigated systematically.
In vivo gene delivery to ocular tissues
Of fundamental importance to all gene therapy strategies is the delivery of genes to specific ocular tissues. Although in vitro, rather than in vivo, delivery is much easier to achieve and may utilise physical techniques (electroporation, microinjection, CaPO4transfection, etc), most of the strategies under investigation for ocular gene therapy require an in vivo approach. The main challenge to date has therefore been to evaluate current vectors for efficient and safe delivery in vivo and how they might be improved.
THE MOUSE MODEL
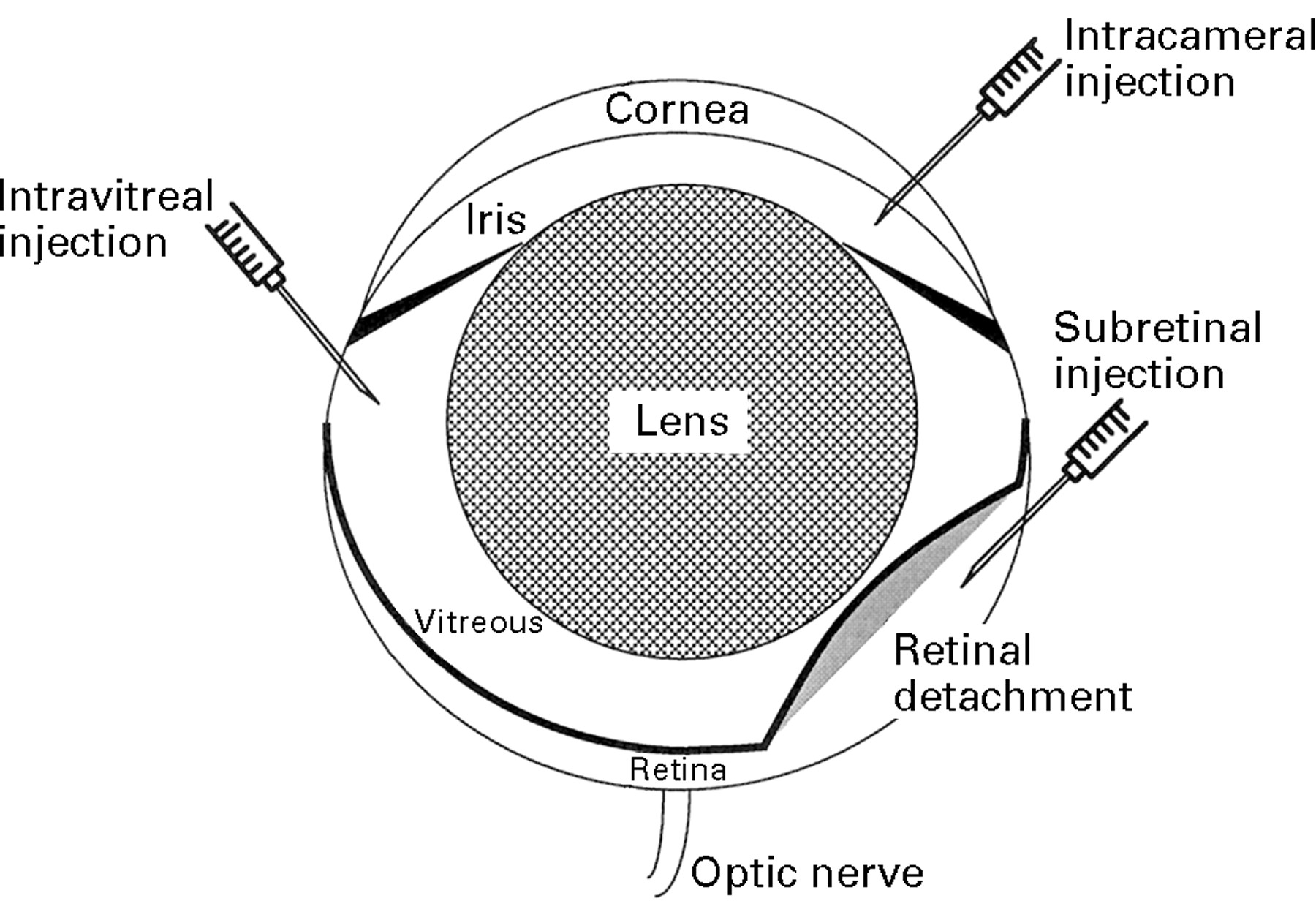
The mouse has been used extensively as an animal model to evaluate gene therapy vectors. This is an appropriate choice since much is known about its biology, and there are many mouse models of ocular disease (comprising spontaneous mutants, transgenic animals, and experimentally induced conditions) which may be utilised for future experiments. It is important to deliver the vectors as close as possible to the target cells. Unfortunately, the small size of the mouse eye, combined with a relatively large lens, makes accurate delivery extremely difficult. Nevertheless, cells in and around the anterior chamber (corneal endothelium, iris pigment epithelium, ciliary body) may be targeted via intracameral injections, ganglion cells via intravitreal injection, and RPE and photoreceptor cells via subretinal injection (Fig1).

Schematic of the mouse eye. Note that the lens is proportionally much larger in mice than in humans. The routes of intraocular injections are indicated.
EVALUATION OF VECTORS
Viruses are little more than highly efficient natural gene delivery systems and are thus obvious vehicles for gene delivery. They have evolved over millions of years to enter mammalian cells (sometimes extremely specifically via receptors) and to avoid endosomal degradation of their RNA or DNA message which is then targeted to the nucleus. The mechanisms by which viruses achieve this have yet to be fully understood. Modified recombinant viruses, which are also replication deficient as a result of deletions in essential viral genes, are currently the most widely used vectors for in vivo gene transfer.1920 Each viral vector system has its advantages and disadvantages and those that have been used to date for ocular gene delivery are reviewed below.
Retroviruses
Retroviruses are ssRNA viruses which require cell division for transfection; recombinant retroviruses are therefore limited in their use for in vivo gene transfer as ocular tissues are either terminally differentiated, or divide very slowly (for example, RPE). Anterior chamber and posterior chamber injections of a disabled (replication deficient) recombinant murine leukaemia virus (rMLV) containing alacZ reporter gene fail to produce transduced cells. This property, however, has been utilised to target dividing cells in brain tumours21 (a strategy which might be also be utilised to target ocular tumours) and more recently to target dividing cells in a rabbit model of proliferative vitreoretinopathy.22 Other potential targets for retroviral mediated gene therapy might be vascular proliferation arising from choroidal or retinal vessels.
Herpes simplex virus (type 1)
Herpes simplex virus (HSV) is a large dsDNA virus. Two types of HSV vector, reviewed by Coffin and Latchman,23 are used for gene transfer—disabled viruses and amplicons (the latter consist of concatamers of plasmids, containing an HSV origin of replication, packaging signal and transgene, packaged into HSV virions by disabled helper virus). We have demonstrated, using a variety of replication deficient HSV vectors containing a lacZ reporter, that this is an effective method of transducing all cell types in the eye, including photoreceptors. The virus has a number of intrinsic properties which make it useful: the potential to accommodate large genes (perhaps up to 30 kb if all non-essential regions are deleted); the possibility of production at high titres, up to 1010 pfu (plaque forming units) (which is useful for ocular transfer where there is a volume restriction) and the ability to remain hidden from the immune system allowing lifelong infections (because it shuts down most viral gene expression during latency). Its natural tropism for peripheral neurons make it of considerable interest for gene delivery to photoreceptor cells.24
However, HSV vectors currently suffer from a number of drawbacks. With most of the currently available vectors, in vivo transduction has been limited to a matter of days. We have investigated several different rHSV vectors and have observed reporter gene expression for up to only 4 days, even in immunodeficient nude animals (unpublished data). The limited duration of reporter gene expression could be due to toxicity of the virus, but is more likely to be the result of the virus entering latency and switching off most gene expression. Finally, wild type HSV, on which recombinant vectors are based, is a highly pathogenic virus. Replication competent HSV, if injected subretinally, will spread along the optic nerve and into the brain where it may cause a fatal encephalopathy (see Fig 2). Thus, maximum consideration must be given to ensuring that rHSV vectors are safe for clinical use. The possibility of rHSV reactivating latent wild type virus in the recipient must be excluded.

Blue X-gal staining indicates lacZ activity in the optic nerve and optic chiasma of a BALB/c mouse 3 days after subretinal injection of 2 μl suspension of replication competent HSV virus, BE8 (5 × 109 pfu/ml) in which the lacZ, driven by a CMV promoter, has been inserted into the non-essential Us5 gene.57
HSV is a complex virus whose biology is still not fully understood, particularly with regard to the mechanism of latency and reactivation. Central to the process of viral inactivation is the latency associated transcript (LAT) which is the only gene that is active during latency. In order to circumvent the switching off of transgene expression during viral inactivation, a number of groups have produced rHSV vectors containing reporter genes driven by the LAT promoter. Lokensgardet al 25 have been able to demonstrate reporter gene expression in mouse dorsal root ganglion cells for up to 42 days. It would be interesting to determine whether similar constructs are capable of driving long term expression in the neuroretina.
Adenovirus
Adenovirus (AV) is a small dsDNA virus. Of all vectors, replication deficient recombinant AV vectors have been the most extensively studied for in vivo gene transfer to the eye.26-33 They have a number of advantages: wild type AV is not highly pathogenic, the vectors may be purified to high titres (1012 pfu/ml), and the virus is able to infect a wide range of ocular cell types.
Anterior chamber injections of AV vectors carrying a lacZ transgene driven by a viral promoter results in the widespread transduction of cells in the anterior segment in mice31 (Fig 3) and rabbits.3233 Corneal endothelium and iris pigment epithelium are readily transduced282931-33 (Fig 4A and 4B). In our experience, the ciliary body, Schlemm’s canal, and trabecular meshwork are also often positive. Although this has been reported by Budenz et al,31 others have not reported transduction of all three structures. This may reflect differences in injection technique or the number of viral particles. The virus does not penetrate the corneal stroma following an intracameral injection and thus does not transduce corneal epithelium. The lens epithelium appears to be resistant to transduction unless the capsule is damaged by the injection.3033 This is more likely to occur during an anterior chamber injection than during an intravitreal injection. Intravitreal injections may result in a similar pattern of transduction, although ganglion cells may now be transduced (Bennett et al reported this in 13% of their intravitreal injections27). Again, variation in the literature may reflect differences in titres and technique. Mashhour et al 29 are alone in reporting transduction of all layers of the retina after intravitreal injection. However, from our own observations this is probably due to an injection technique which disrupts the retina.
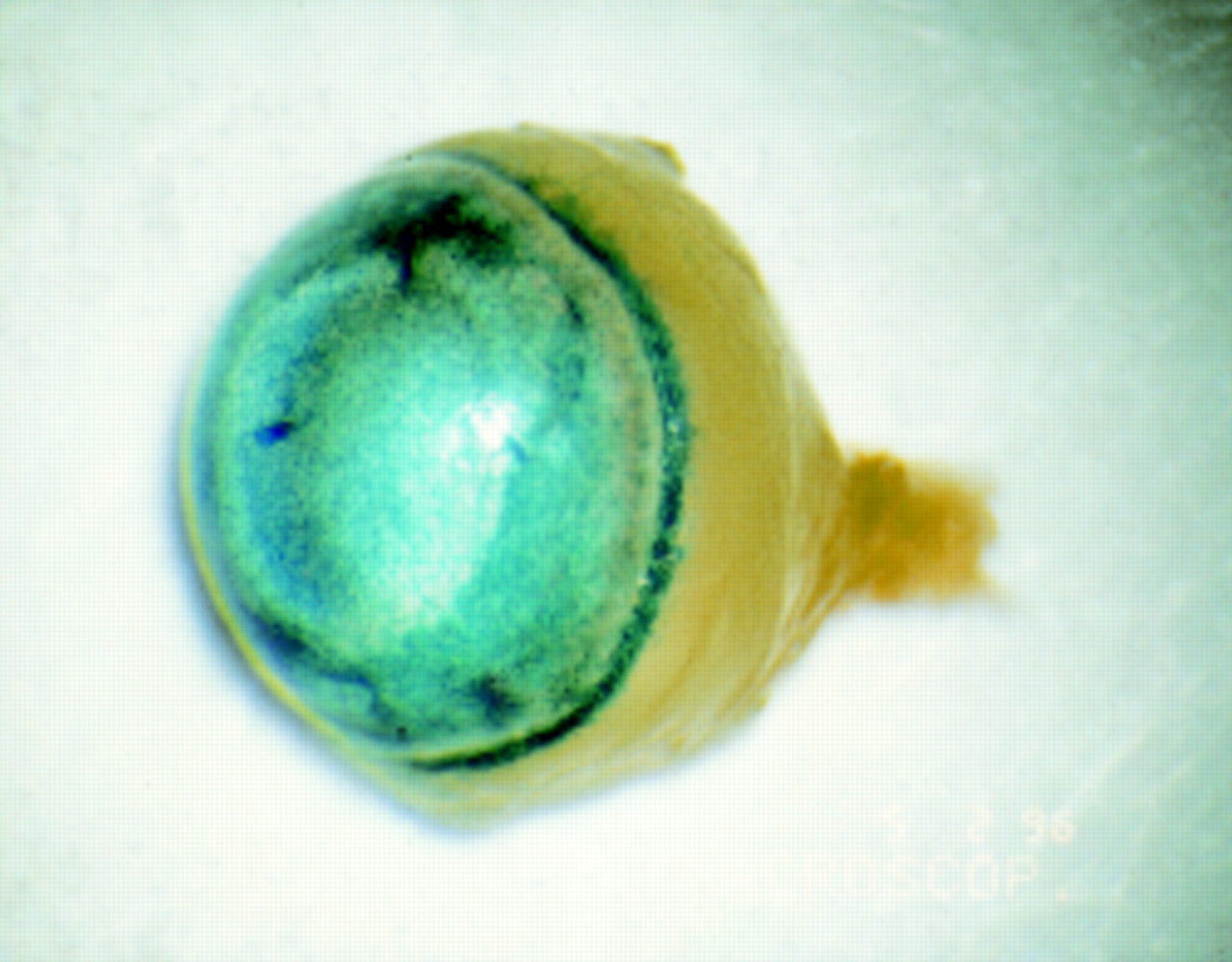
Blue X-gal staining in the anterior chamber of adult BALB/c mouse after intracameral injection of 2 μl of adenovirus carrying a lacZ gene with nuclear localisation signal driven by a CMV promoter (AV.CMV.LacZnuc) at a titre of 1 × 109 pfu/ml. LacZ activity can be observed throughout the anterior segment including corneal endothelium, iris, and trabecular meshwork.

Transduced corneal endothelium (A) and iris pigment epithelium (B) in adult BALB/c mouse after intracameral injection of 2 μl of AV.CMV.LacZnuc (1 × 109 pfu/ml). A 5 μm paraffin section counterstained with nuclear fast red (× 45).
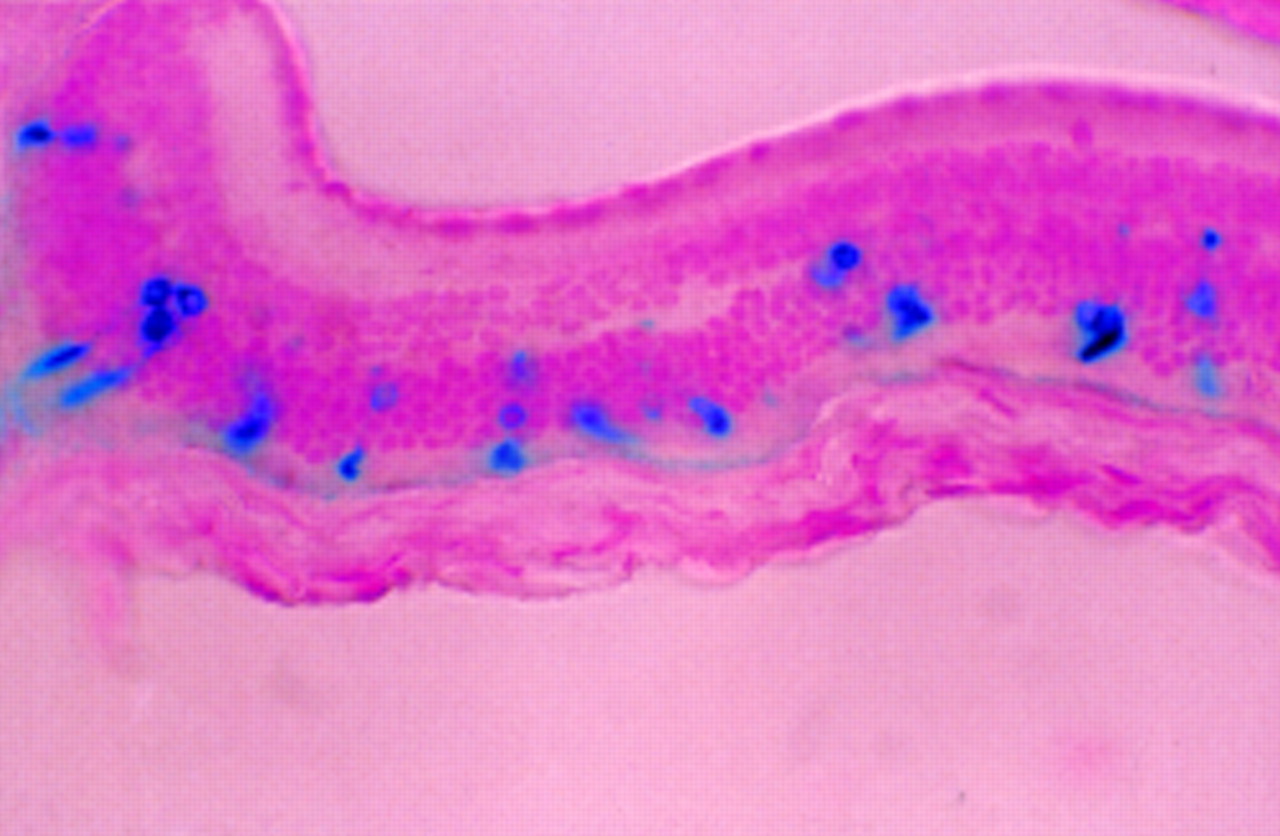
After subretinal injection, highly efficient transduction of RPE is observed but there is very poor transduction of photoreceptor cells272834 (Fig 5). These cells may, however, be more efficiently transduced if they are in the process of development (neonatal mice at day 5) or in the process of degeneration (2.5-month-old rds mice).28 In both cases, photoreceptors may be more accessible to virus. It is unlikely that the first observation is of clinical relevance for treating inherited retinal degeneration since human photoreceptors are fully developed at birth. However, the second is worth noting since the first clinical trials would probably take place in older patients, where a higher transduction efficiency may offset problems associated with the treatment of degenerate retinas. Higher titres of AV result in much better transduction of photoreceptors. Bennett et alreported that in adult mice transduction of the neuroretina required the injection of at least 1 × 107pfu.27 However, this must be weighed against the possibility of direct toxicity to the cells, especially to adjacent RPE cells which take up the virus very efficiently. One week after subretinal injection, disruption of the retina and damage to photoreceptor outer segments can be observed,2728 but by 6 weeks this is resolved.28 Cytopathic changes have been observed after intraocular injection of 108 pfu in mice.28 At 8 weeks post-injection, Li et alobserved that there were a reduced number of photoreceptors in some sections.

Transduced retinal pigment epithelium and occasional positive photoreceptor cell (arrow) in adult BALB/c mouse 2 weeks after subretinal injection of 2 μl of AV.CMV.LacZnuc (1 × 109pfu/ml). A 5 μm paraffin section counterstained with nuclear fast red (× 66).
In our experience, using BALB/c mice, ocular AV mediated reporter gene expression usually lasts no more than 3 weeks (Ali, unpublished results). We have also demonstrated that an immune response limits the duration of expression in both anterior and posterior segments and is directed primarily at the vector (manuscript in preparation). In general, inflammatory cells have not been noted in post-injected retinas. However, they may be present in the vitreous which is usually removed during processing. Borras et al 33have observed infiltrating cells in rabbits 2 days after anterior chamber injection of 109 pfu. It appears therefore that ‘immune privilege’ associated particularly with transplants into the anterior chamber (but more recently also to the subretinal space35) does not apply to cells infected with virus by intraocular injection. This is perhaps not surprising since virus injected into the anterior segment drains out into the venous blood stream and injection of virus into the posterior segment disrupts the blood-retinal barrier. Li et al,28 however, have observed scattered reporter gene expression 6 weeks after injecting neonatal CB-17 mice (which are almost genetically identical to BALB/c), presumably by circumventing the developing immune system. Although there have been reports of much longer gene expression after intraocular injection of adult mice (for 13 weeks in two CD-1 mice27) strain differences may account for some of the discrepancies. We have shown, for instance, that lacZ expression is maintained for around 1 week longer in MF1 mice than in BALB/c mice. The immunogenicity of AV vectors may be reduced in the future as new vectors are being developed which contain fewer viral genes. This may, however, generate a new problem of efficient in vitro production of recombinant virus. Alternatively it may be possible to tolerise animals to the vector.
Adeno-associated virus
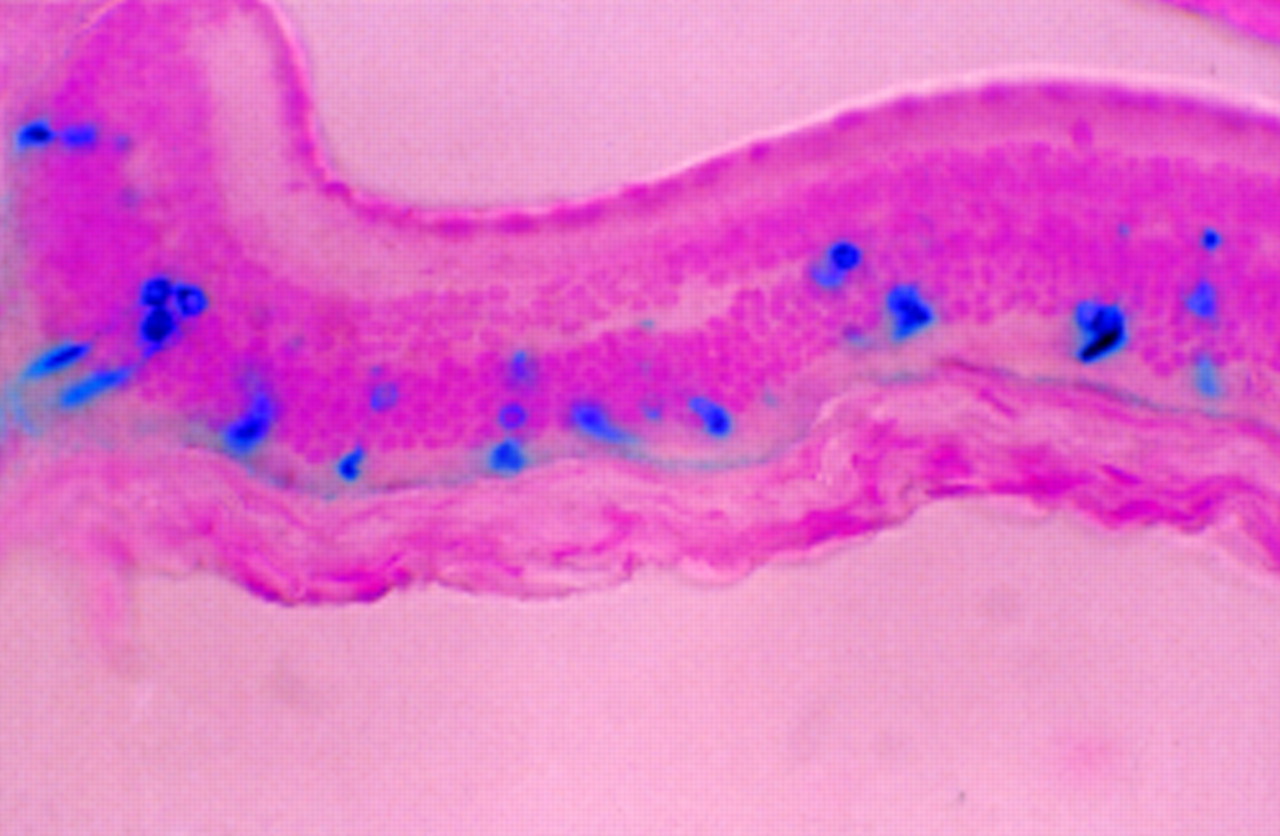
Adeno-associated viral (AAV) vectors have generated considerable interest recently. They are potentially very attractive since wild type AAV, a tiny ssDNA parvovirus, is not associated with any pathology in humans (40% of the population is seropositive for AAV without ill effect). In addition, recombinant AAV vectors are deleted for all virally encoded proteins, therefore reducing their immunogenicity. Furthermore, rAAV is capable of transducing non-dividing neuronal cells. Although the wild type virus integrates site specifically into chromosome 19, rAAV lacks the ability to do this and probably remains in the nucleus as an episome. We have now demonstrated that, after subretinal injection, rAAV is 2000-fold more efficient at transducing photoreceptor cells than a rAV vector containing a similar reporter construct34 (Fig 6). As with AV, the RPE is very efficiently transduced, and we have also observed transduced cells in the bipolar and ganglion cell layers. Expression of the reporter gene in immunodeficient mice was detected up to 1 month after injection.34 Because of its small size, the virus may be able to penetrate the layers of the retina. After subretinal injection of mice we have observed transduced ganglion and bipolar cells and Zolotukhin et al have observed transduced RPE cells in the guinea pig after intravitreal injection of a rAAV carrying a green fluorescent protein (GFP) reporter.36
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Transduced photoreceptor cells in adult nude mouse 1 month after subretinal injection of 2 μl of AAV.CMV.LacZ (1 × 107 IU/ml). All the stained outer segments can be related to stained photoreceptor nuclei which is consistent with transduction of photoreceptor cells and subsequent transport of LacZ into the outer segments. A 5 μm paraffin section counterstained with nuclear fast red (×25).
AAV vectors currently have a number of limitations: the virus is very difficult to prepare to high titres and without contamination with helper AV. The maximum size of insert that the vector is able to accommodate is only 4.7 kb. In addition, it now appears that transduction by AAV may require coinfection of cells with (contaminating) wild type AV which may facilitate the conversion of ssDNA to the ds form.3738 These problems will need to be addressed before AAV vectors can be considered as suitable vectors for gene delivery in humans.
Future vector development
The DNA viruses which have been investigated to date (HSV, AV, and AAV) are all efficient at transducing non-neuronal cells, in particular the RPE, which probably phagocytoses the virus. A major challenge for ocular gene therapy is to further improve the efficiency of transduction of neuronal cells and in particular photoreceptor cells, and to reduce the immune responses in order to achieve long term gene expression. Each of the viral vectors have potential advantages and disadvantages, but in general it appears that AV vectors may be most suitable for transducing non-neuronal ocular cells, whereas AAV appears to be a promising vector system for gene delivery to photoreceptor cells—provided the requirement for wild type AV can be overcome. HSV vectors probably require the most development.
Although a number of vectors have already been used for ocular gene delivery, it will be essential to evaluate new vector systems as they are developed. For instance, the possibilities of utilising modified lentiviruses for gene transfer to photoreceptors has recently been raised with the demonstration of reporter gene delivery to neuronal cells using an HIV vector.39 The potential advantage of using such retroviruses is the possibility of stable transduction. Alternative vector systems which utilise a viral packaging system, but contain minimal viral DNA (such as HSV amplicons and the polyoma system) have not yet been investigated with respect to their potential use in the eye. Although non-viral gene delivery systems such as liposomes have so far proved to be generally very inefficient in vivo (also in the eye, unpublished data), they have the advantages of low toxicity and immunogenicity and with further development may prove useful. They may also be used to improve the transduction efficiency of viral vectors.40
Experimental gene therapy strategies using mouse models of retinal degeneration
An essential requirement for the development of gene therapy is the availability of suitable well characterised animal models. Since mice are relatively inexpensive to maintain, well characterised genetically, and are used for transgenic manipulations most animal models are murine (and given the rapid progress in mouse genetics this will continue to be so in the future). Various gene therapy strategies can be employed to treat disease in these models. In general, ablation of specific cells is often the goal for gene therapy for experimentally induced non-genetic diseases such as uveal melanoma and proliferative vitreoretinopathy (and is outside the scope of this review), whereas restoration of function is the goal for gene therapy of genetic disorders.
GENE DELIVERY TO PHOTORECEPTORS
Recessive disorders, caused by loss of function, and those dominant diseases caused by haploinsufficiency, where 50% of the gene product is inadequate for normal function, are more straightforward to treat by gene therapy than dominant disorders in which there is a gain of function. The remainder of this section concentrates on the two extensively characterised mouse strains, rd (retinal degeneration) and rds (retinal degeneration slow), which respectively provide recessive and dominant haploinsufficiency models of RP. In each case the photoreceptor cells degenerate and are lost through apoptosis.41
The defect in the rd mouse is a recessive null mutation in the bPDE gene (which encodes the β subunit of rod phosphodiesterase) and leads to a rapid and severe degeneration in homozygous mice.42 Photoreceptors are lost within 1 week of birth, with 50% loss by 2 weeks (heterozygous mice are indistinguishable from wild type). Bennett et al have slowed the degeneration in the rd mouse by subretinal injection in 5-day-old pups of a rAV vector carrying a bPDE gene driven by a viral promoter.18 Three weeks after injection, they were able to demonstrate areas of the retina with three or four layers of photoreceptor cells (compared with one layer in the untreatedrd mouse and eight to nine layers in a normal adult mouse). By 7 weeks there was little difference between treated and untreated mice. It is not clear what proportion of photoreceptors had been transduced but it seems unlikely that the additional layers of outer nuclei consist only of transduced cells. It is possible that this phenotype resulted from the transduction of scattered photoreceptors as might be expected from previous studies,2728 with rescue of surrounding cells. Alternatively, rescue may have been effected primarily by transduction of RPE cells with secretion of the βPDE gene product and uptake by the photoreceptors. These possibilities could be readily distinguished by immunohistochemical staining for βPDE and by repeating the experiment using a rAV carrying the βPDE gene driven by a photoreceptor specific promoter. The defect in therds mouse is a null mutation in the peripherin/rds gene which encodes a structural protein required for outer segment stability.43 These mice were first characterised by Sanyalet al.44 Mice homozygous for therds mutation lack outer segments and 3 weeks after birth begin to lose photoreceptor cells. By 2 months, the outer nuclear layer is reduced to approximately half its thickness. Mice heterozygous for the mutation are characterised by abnormal outer segments and a much slower loss of photoreceptor cells.45 The defect is thus more accurately described as a semidominant mutation since the heterozygote has an intermediate phenotype between wild type and homozygous mice and is due to haploinsufficiency. The heterozygousrds mouse will prove particularly useful for gene delivery to photoreceptor cells since the pathology is rather mild and the rate of degeneration is relatively slow, making it a good model for late onset autosomal dominant RP. Once photoreceptor cells have degenerated beyond a certain point, gene delivery is unlikely to be beneficial. Since the peripherin/rds protein is a structural protein, overexpression of the gene in photoreceptor cells may be deleterious: the photoreceptors of transgenic mice carrying multiple copies of a normal rhodopsin transgene eventually degenerate.46Therefore, long term correction of the defect in this mouse model will probably require precise control of the level of expression of the introduced gene—with appropriate promoter and multiplicity of infection.
There are a number of transgenic mouse models of dominant RP, in which photoreceptor degeneration results from dominant mutations in either a rhodopsin46-48 or peripherin/rds transgene (G Travis, personal communication). One strategy to slow degeneration in these mice might be to overexpress the normal gene, thereby competing out abnormal protein with normal. However, it is unclear what ratio of proteins might be required and to what extent overexpression of the normal protein would lead in itself to disease. Prevention of the degeneration will probably require specific inactivation of the abnormal message. In the case of dominant rhodopsin mutations this should be sufficient to prevent disease since there would be haplosufficiency (individuals who are heterozygous for a rhodopsin null mutation are unaffected49). However, in the case of dominant peripherin mutations, where there is a gain of function, inactivation of the abnormal message may not be entirely sufficient to prevent disease since this may then lead to haploinsufficiency. The potential of ribozymes has been demonstrated in vitro and that of antisense DNA in cell culture. Recent evidence has supported the use of antisense constructs in vivo.50-52
Initially, it is simpler to assess the effects of gene transfer on correcting abnormal ultrastructure and histology, than to assess its effects on visual transduction. Since Huang et al have demonstrated, with aggregation chimeras between normal and transgenic mice expressing a mutant rhodopsin gene, that normal photoreceptors degenerate if surrounded by abnormal photoreceptors,48 it may not be sufficient to rescue a small proportion of cells. Because photoreceptor cells are so difficult to target, there is still much work left to be done. Our best efforts to date have resulted in transduction of less than 1% of the total photoreceptor cell population.
GENE DELIVERY TO THE RPE
Transduction of the RPE is much more efficient than that of neuroretina, probably reflecting the phagocytic property of this cell type. Primary biochemical defects in the RPE may lead to photoreceptor degeneration. For instance, the retinal dystrophy (rdy) rat (also known as the Royal College of Surgeons’ rat) has a single, as yet unidentified, gene defect of the RPE which results in retinal degeneration.53 It is tempting to speculate that a proportion of the uncloned RP genes may turn out to have a function in the RPE. These diseases would thus be prime candidates for gene therapy (and indeed the candidate gene for RP3, RPGR, may prove to be such a gene54). Age related macular degeneration (AMD), which has both environmental as well as genetic components, is also thought to result partly from an RPE abnormality (accumulation of lipofuscin). This widespread condition is also a potential candidate for gene therapy. Overexpression in the RPE of enzymes which degrade lipofuscin deposits may ameliorate the condition. Although an accurate model of AMD does not exist, the mnd mouse, a model for Battens’ disease, exhibits a build up of lipofuscin in the RPE55and might be useful for testing such therapies.
An alternative strategy to treat retinal degeneration might involve the use of growth factors to promote photoreceptor survival. This approach, using either direct injection or recombinant viruses to deliver brain derived neurotrophic factor (BDNF) and ciliary neurotrophic factor (CNTF) to the central nervous system, has had some success in reducing the damage seen in several neurodegenerative mouse models.56 Although single intravitreal injections of either BDNF or CNTF into rd and rds mice have not slowed the degeneration, it is possible that long term treatment with these factors may ameliorate the condition. Given the efficiency with which the RPE and corneal endothelium may be transduced, this might be achieved using either rAV or rAAV to deliver the various growth factor genes to these tissues.
A number of neurodegenerative diseases have an associated retinal degeneration. One such disease is mucopolysaccharidosis VII, a recessive lysosomal storage disease resulting from a deficiency of β glucuronidase, that leads to lysosomal accumulation of glycosaminoglycans in the RPE and subsequent photoreceptor degeneration. Li and Davidson have reported the phenotypic correction of the RPE defect in the gusmps murine model of mucopolysaccharidosis VII using a rAV vector.30However, it is not yet clear whether this resulted from replacing the missing gene product since, in the absence of a suitable control (for example, rAV carrying lacZ), it is also possible that the transduced RPE cells had been ablated by a cytotoxic immune response to the vector, and then been replaced by new RPE cells. These new cells may then not have had sufficient time for the lysosomal accumulation of glycosaminoglycans.
Conclusion
The potential for gene therapy in the treatment of ocular disease has only recently been explored. Although in vivo gene delivery of reporter genes, using a variety of viral vectors, has indicated the feasibility of ocular gene therapy, considerable improvements with respect to efficiency of the currently available vector systems is required. In particular, the efficiency of transduction of photoreceptor cells must be improved and the duration of expression increased. The problem of immune responses to exogenous proteins (particularly derived from the vector) should be addressed since, as in other organs, they will limit the duration of expression of transgenes in the eye. Substantial progress in the treatment of disease in animal models must be demonstrated, and concerns regarding the safety of vectors satisfied, before clinical trials can be contemplated. However, past experience justifies a cautious optimism that considerable improvements to this emerging technology will be achieved in the next decade.