Article Text

Statistics from Altmetric.com
Sarcoidosis is a chronic multisystemic granulomatous disorder thought to result from an exaggerated cellular immune response to a variety of self antigens or non-self antigens.1 The aetiology of sarcoidosis is unknown, which might be in part related to the diverse manifestations of the disease and the absence of approved diagnostic criteria. Although specific inhalation antigens have been put forward as possible triggers, no study has yet proved a consistent relation with a causative agent.2 Genetic factors might also be involved since familial incidence in certain populations as high as 19% has been noted and, further, specific HLA associations have been connected with disease susceptibility and outcome—for example, HLA-B8 and DR-3 with Löfgren's syndrome (acute systemic disease with fever, polyarthritis, erythema nodosum, and favourable prognosis).3 4 The infectious agents, specifically mycobacteria and more recently herpesvirus 8, have also been implicated.5 6 Sarcoidosis has only sporadically been reported in AIDS and has occasionally been described during the immune recovery with the highly active antiretroviral therapy.7 8 The evidence that disease might be of environmental or infectious origin is supported by occupational clustering (medical nurses, fire fighters) and by the transmission of the disease from transplanted organs.9-11 The genetic and environmental influences cannot be adequately evaluated in small samples of patients; a large study addressing the phenotyping differences and various environmental data is lacking however. So far, only interesting disease patterns in families and inconclusive allelic associations for different populations have been reported. Therefore, sarcoidosis might represent an abnormal immune response directed against one or more possible antigens in an individual with a hereditary or acquired abnormality of the immune system.
Epidemiology
Sarcoidosis occurs worldwide but is predominant in certain ethnic and racial groups (for example, US blacks, Scandinavian and Irish white people).12 Black patients present typically with more severe and acute disease whereas the white patients usually have asymptomatic and chronic disease.13 14 The frequency of sarcoidosis is reported to be low in various parts of the world14 15; it is not known whether this low frequency of sarcoidosis is genuine or whether it represents an underdiagnosis owing to the frequent occurrence of subclinical course, similarity with other diseases, or absence of firm diagnostic criteria.
Pathogenesis
Sarcoidosis is characterised by the formation of non-caseating granulomas in affected organs. Three types of agents are known to cause epitheloid cell granulomas—infectious organisms (bacteria and fungi), products of plants and animals (pollen, proteins), and metallic compounds (beryllium, zirconium). In sarcoid granulomas, the presence of mycobacterial DNA was identified by molecular techniques and a causal role in the aetiology of sarcoidosis was proposed.16 17 Conflicting results have been obtained from the studies assessing the aetiological role of mycobacterial infection in the pathogenesis of sarcoidosis and the discrepancies were attributed to different sensitivities of the diagnostic procedures.18 19 The nested polymerase chain reaction (PCR), a procedure more sensitive than the standard PCR protocol, has failed to identify sequences specific forMycobacterium tuberculosis complex in sarcoidosis, whereas positive results were found in biopsies from patients with active tuberculosis.20 21 Polymerase chain reactions of 123 bp fragment of IS6110 have shown the absence ofM tuberculosis DNA in lymph nodes and lung biopsies from patients with sarcoidosis and suggest thatM tuberculosis does not have a pathogenic role in sarcoidosis in most patients.22 By molecular techniques, atypical mycobacteria, Corynebacterium acnes, and Propionibacterium DNA have also been identified in sarcoid granulomas and a link with sarcoidosis has been proposed.23 24 Whether these bacteria induce some cases of sarcoidosis by an allergic mechanism has not yet been elucidated.
Presumably, in response to antigen exposure, exuberant cellular immune response occurs in target organs, and leads to the development of granulomas. Granulomas consist of epitheloid and multinucleated giant cells (aggregated macrophages), abundant CD4 positive T cells and, usually in the periphery of the granuloma, to some extent CD8 positive T cells and B cells may be encountered.25 26 T lymphocytes in affected organs are of T helper 1 phenotype and produce interferon γ and interleukin 2, which subsequently lead to the production of tumour necrosis factor and interleukin 6 by macrophages, and thereby cause a cascade of inflammatory changes, culminating in fibrosis. Fibrosis of the granulomas induces organ destruction and loss of function. The very high ratios of T helper to T suppresser cells have been reported for various organs affected by sarcoidosis, including the eye.26 Epitheloid cell granulomas are not pathognomonic for sarcoidosis, and other causes of granulomatous inflammation (for example, Wegener's disease) as well as the presence of infectious agents (fungi or mycobacteria) and foreign bodies (beryllium, organic dusts) should be sought.1 Probably secondary laboratory abnormalities include depressed cellular immunity to other antigens (resulting in cutaneous anergy), hyperglobulinaemia caused by overstimulated B lymphocytes, and hypercalcaemia.
So far, the search for the associations between HLA related genes and sarcoidosis has revealed that the most common allele associated with the susceptibility to sarcoidosis has been HLA-B8.27 The present studies, using the DNA sequence analysis rather than serological typing, support the hypothesis that the major genetic factors controlling the development of sarcoidosis are located within the DRB1 locus in the HLA class II region.28 29 Several studies on genetic polymorphism have shown the potential to predict the clinical course of the disease.28 Although the HLA class II genotyping was associated with disease outcome in Scandinavian patients (HLA-DR 14 and 15 were associated with chronic sarcoidosis and HLA-DR 17 with non-chronic forms of sarcoidosis), in other populations other alleles were responsible for the susceptibility and course of the disease.30 31
Clinical manifestations
The course of sarcoidosis ranges from asymptomatic to severe and even lethal disease. The clinical manifestations and severity of the disease vary widely and are strongly associated with racial and ethnic factors: acute and more severe disease is typical of black patients, whereas asymptomatic and chronic disease is more frequent in whites.12 In all races, however, mortality remains similar.32 Although the overall prognosis is relatively benign, many patients develop some organ dysfunction. Factors associated with poor outcome include black race, late onset, and disease persisting for longer than 6 months, involvement of more than three organs, and stage III of pulmonary disease (parenchymal infiltrates without adenopathy).1 33 Sarcoidosis affects people of all ages with the peak age of onset in the third decade.13
The disease affects predominantly the lungs and thoracic lymph nodes, skin, and eyes and might have either self limited or chronic course. Although the manifestations of sarcoidosis may be widespread, as a result of the frequent manifestations in lungs and lymph nodes, most patients will have some respiratory problems.13 The majority of patients have constitutional symptoms such as fever, malaise, fatigue, and weight loss. Frequently the disease is asymptomatic and discovered by chest radiography (hilar adenopathy) performed for unrelated causes. The appearance of the chest radiograph is usually used to classify a pulmonary sarcoid: stage 1 represents bilateral hilar adenopathy without parenchymal involvement, stage 2 bilateral adenopathy with parenchymal involvement; and stage 3 is pulmonary infiltrates without hilar adenopathy, including cystic changes.34 35 Specific manifestations of sarcoidosis include acute Löfgren's syndrome, which is a combination of erythema nodosum, arthritis, and hilar lymphadenopathy (sometimes associated with anterior uveitis) and Heerfordt's syndrome (uveoparotid fever), consisting of fever, parotid swelling, uveitis, and sometimes facial palsy.36 Most frequently affected extrapulmonary organs are lymph nodes, eyes, and skin. Skin lesions are common and include, in addition to sarcoid granulomas, erythema nodosum. Erythema nodosum represents a reaction pattern of the skin to diverse causes (the spontaneously healing lesions are tender red nodules located usually on the lower extremities); the lesions are not granulomatous and therefore biopsy does not contribute to the diagnosis of sarcoidosis. The development of granulomas is regularly noted in scar tissues. CNS manifestations occur in 5%–12% of patients.1 Renal insufficiency may develop secondarily to granulomatous nephritis or hypercalcaemia. Cor pulmonale may develop secondarily to severe pulmonary disease. Direct sarcoid infiltration of the heart muscle is seen in about 5% of patients with sarcoidosis.
Sarcoidosis in children is frequently of extrapulmonary type with frequent ocular as well as cutaneous and rheumatological manifestations and has a guarded prognosis.37 38 The triad—rash, uveitis, and arthritis—is characteristic for children under 4 years of age; more frequent is the occurrence of sarcoidosis in older children (8–15 years of age), which is characterised by almost universal lung involvement and manifestations in eyes, skin, liver, and spleen. Long term follow up illustrated a high frequency of blindness and severe multisystem (for example, heart and renal) involvement in children with onset of sarcoidosis at a very early age.37 Childhood sarcoidosis might be misdiagnosed because of its rarity and resemblance to juvenile chronic arthritis. Autosomal, dominantly inherited Blau syndrome (familial granulomatous inflammatory arthritis, uveitis and rash, and camptodactylia) is characterised by multiorgan inflammation and may strongly resemble sarcoidosis; however, the typical pulmonary involvement is absent.39-41
Diagnostic methods
In the absence of the known causative agent, the diagnosis of sarcoidosis remains mainly a diagnosis of exclusion. Since there are no definitive diagnostic serological or radiological tests, the presence of non-caseating granulomas on tissue biopsy together with compatible clinical features is usually considered as a proof of the diagnosis of sarcoidosis. However, even in cases with tissue biopsy consistent with the presence of sarcoidosis, other possible causes of granulomatous diseases should be evaluated. Although biopsy is not pathognomonic, tissue examination is essential to differentiate sarcoidosis from infections or malignancies. Tissue biopsy might be obtained from lung, skin, lymph node, or other affected organs. Undirected biopsies yielded less positive results compared with those obtained from overtly affected organs; this was also reported for conjunctival granuloma or lacrimal gland biopsy.42-44 The analysis of fluids and cells obtained by bronchoalveolar lavage, including CD4/CD8 ratio, is not specific enough for the definitive diagnosis of sarcoidosis.45 Chest x ray usually shows hilar adenopathy in combination with interstitial lung disorder. Chest computed tomography has been reported to be more sensitive than radiography.46 Measurements of serum angiotensin converting enzyme (ACE) level reflect the disease activity; ACE levels fluctuate with corticosteroid use, but are not specific enough for diagnostic purposes.47 48 High levels of serum ACE were also reported for other diseases (for example, tuberculosis, leprosy, diabetes mellitus). Moreover, as serum ACE levels reflect the systemic burden of inflammation, normal serum ACE levels do not exclude the diagnosis of sarcoidosis, especially not in those with isolated ocular disease. The patients with presumed sarcoidosis (intraocular inflammation compatible with the diagnosis of sarcoidosis in conjunction with raised serum ACE levels) were similar to those with histologically proved sarcoidosis in terms of ocular presentations and clinical course.49 The combination of raised serum ACE levels with abnormal gallium scanning was a specific and sensitive tool for diagnosing patients suspected of having ocular sarcoidosis who had normal chest radiographs.50 Aqueous ACE levels of patients with sarcoid associated uveitis were increased, though their diagnostic value was not yet determined.51 Increased levels of ACE in cerebrospinal fluid were reported, but their diagnostic role is not clear.52 Gallium scan uptake, serum lysozyme levels, hypergammaglobulinaemia, and decreased delayed type hypersensitivity are not specific for sarcoidosis. Kveim–Siltzbach test, which uses human tissue obtained from patients with sarcoidosis to induce granuloma in a tested patient, has been abandoned. Although lung involvement occurs at some time in the course of the disease in nearly all cases with sarcoidosis, the sensitivities and specificities of various diagnostic procedures for patients with ocular sarcoidosis are not known. In patients with uveitis, sensitivity of raised serum ACE was 73% and specificity 83%.50 In patients with active systemic sarcoidosis, gallium-67 scanning had a sensitivity of 95%, but a low specificity (68%), while the sensitivity of chest radiography was 80%, and of serum ACE 77%.53
Treatment
The optimal therapy for sarcoidosis is not well defined; therapeutic decisions are dictated by the localisation of the disease and severity of organ involvement. The mainstay of treatment is corticosteroid therapy, which exhibits especially short term beneficial effects.34 The controversy remains, however, concerning the efficacy of corticosteroids to alter the natural course of the disease.35 36 54 For refractory disease, methotrexate was reported to be the most promising drug and was recently reported to be effective in ocular involvement.54-56 The efficacy of antimalarial agents and cyclosporin has not been conclusively proved.57-59
Ocular involvement
Ocular disease may be the initial manifestation in sarcoidosis and may progress to severe visual impairment or even blindness. No specific extraocular manifestations of sarcoidosis were associated with the development of ocular involvement or uveitis.60 61 Two peaks of incidence were reported for ocular sarcoidosis, the first at ages 20–30 years and the second at ages 50–60 years.61Ocular involvement manifests in 25%–60% of patients with systemic sarcoidosis.62 This percentage depends on the population studied and the extent of ocular examinations. The most common ocular manifestations are uveitis (30%–70%) and conjunctival nodules (40%).62-66
Uveitis is a frequent and early feature of sarcoidosis. More than 80% of uveitis cases manifested before or within 1 year after the onset of systemic disease. Uveitis preceded the non-ocular signs of sarcoidosis in about 30%.61 In large surveys of uveitis patients, the frequency of sarcoidosis was about 5%.62Traditionally, the most common type of sarcoid associated uveitis was attributed to anterior uveitis. It is noteworthy that the high frequency of sarcoid associated anterior uveitis (70%–75%) was noted mainly in the studies where the majority of patients were black.62 63 66-68 In contrast, posterior type was the most common localisation of sarcoid uveitis in white patients (65%–83%), specifically in elderly female patients.49 61 62 65 69 The visual impact of sarcoid associated uveitis was severe, about 10% of patients developed blindness in at least one eye.70 The main cause of visual loss was cystoid macular oedema.49 71 The poor visual prognosis was associated with the advanced age of the patients, black race, female sex, chronic systemic disease, and also with posterior segment involvement, peripheral punched out lesions, and the presence of cystoid macular oedema and glaucoma.69 71-73
CLINICAL MANIFESTATIONS
Neurological manifestations of sarcoidosis are varied and were identified in 12% (71/554) of patients with sarcoidosis. Posterior segment eye involvement was frequently accompanied by CNS involvement.62 Neurological manifestations may include optic nerve disease, cranial nerve palsies, encephalopathy, and disorders of the hypothalamus and pituitary gland.74 75Chiasmal syndromes and motility disorders may also occur. Facial nerve palsy was the most common but usually self limited manifestation of CNS involvement. Optic nerve involvement may be caused either by direct sarcoid tissue infiltration or compression by cerebral mass, and may lead to optic nerve atrophy.76-78 Usually, the involvement of the optic nerve is an indication for systemic treatment. The treatment of chronic neurological symptoms with methotrexate or cyclophosphamide was associated with a better therapeutic response than the treatment with corticosteroids alone.
Lids and orbit
Lacrimal gland or conjunctival involvement is common and usually asymptomatic, although extensive granulomas leading to diplopia or severe keratoconjunctivitis sicca may develop.79 80Eyelid or conjunctival granulomas were repeatedly reported and usually react well to topical treatment.81 In resistant cases, an intralesional corticosteroid administration may help. Other manifestations in orbit and lids are not frequent and are usually unilateral.82 Orbital symptoms mimic other inflammatory syndromes manifesting in the orbit. Sarcoid induced inflammatory myositis may resemble Graves' ophthalmopathy. The association of sarcoidosis with autoimmune thyroid disease is well known and the coincidence of sarcoidosis with Graves' disease has also been noted.83
Corneal involvement by granulomas is extremely rare.84Corneal band degeneration may develop as a consequence of long standing anterior uveitis.
Uveitis
Acute signs of uveitis as pain, photophobia, lacrimation, or redness might be absent, so that the patients with sarcoid associated “silent uveitis” may develop permanent ocular damage before the intraocular process is diagnosed and treatment initiated. In a series of 121 patients with biopsy proved sarcoidosis, 29 patients were diagnosed with uveitis. Of these 29, 10 were diagnosed by an ophthalmologist in the absence of ocular complaints at the time of initial ophthalmological screening.61
Classic sarcoid associated anterior uveitis may either present as acute iridocyclitis, which is mostly seen in Löfgren's syndrome, or as a chronic granulomatous uveitis with keratic precipitates, which may vary from cellular to large “mutton fat” type. In the chronic type of the disease, the granulomatous nodules of the iris (Fig 1A) and in the anterior chamber angle are sometimes seen. These are usually whitish and range in size from small to large masses (Fig 1B). Granulomas located on trabecular meshwork (sometimes associated with elevated eye pressure) were noted in 49% of Japanese patients.85Chronic anterior uveitis may lead to secondary cataract, glaucoma, and cystoid macular oedema; corneal band keratopathy may also develop.

Anterior segment granulomas in sarcoidosis. (A) Granulomatous iridocyclitis in sarcoidosis with Koeppe nodules visible on the pupillary border. (B) Multiple, extremely large granulomas in anterior chamber in a black patient with biopsy proved sarcoidosis. Keratic precipitates located on the lower part of corneal endothelium are also visible.
Intermediate uveitis with vitritis and genuine snow banking may be occasionally encountered. More frequent is the presence of vitritis with peripheral vasculitis and snowball infiltrates. This type of intermediate uveitis may precede more severe posterior segment changes. Snowball opacities in the vitreous cavity are characteristic but not exclusive for sarcoidosis and the differentiation from idiopathic pars planitis and other types of intermediate uveitis might be difficult.
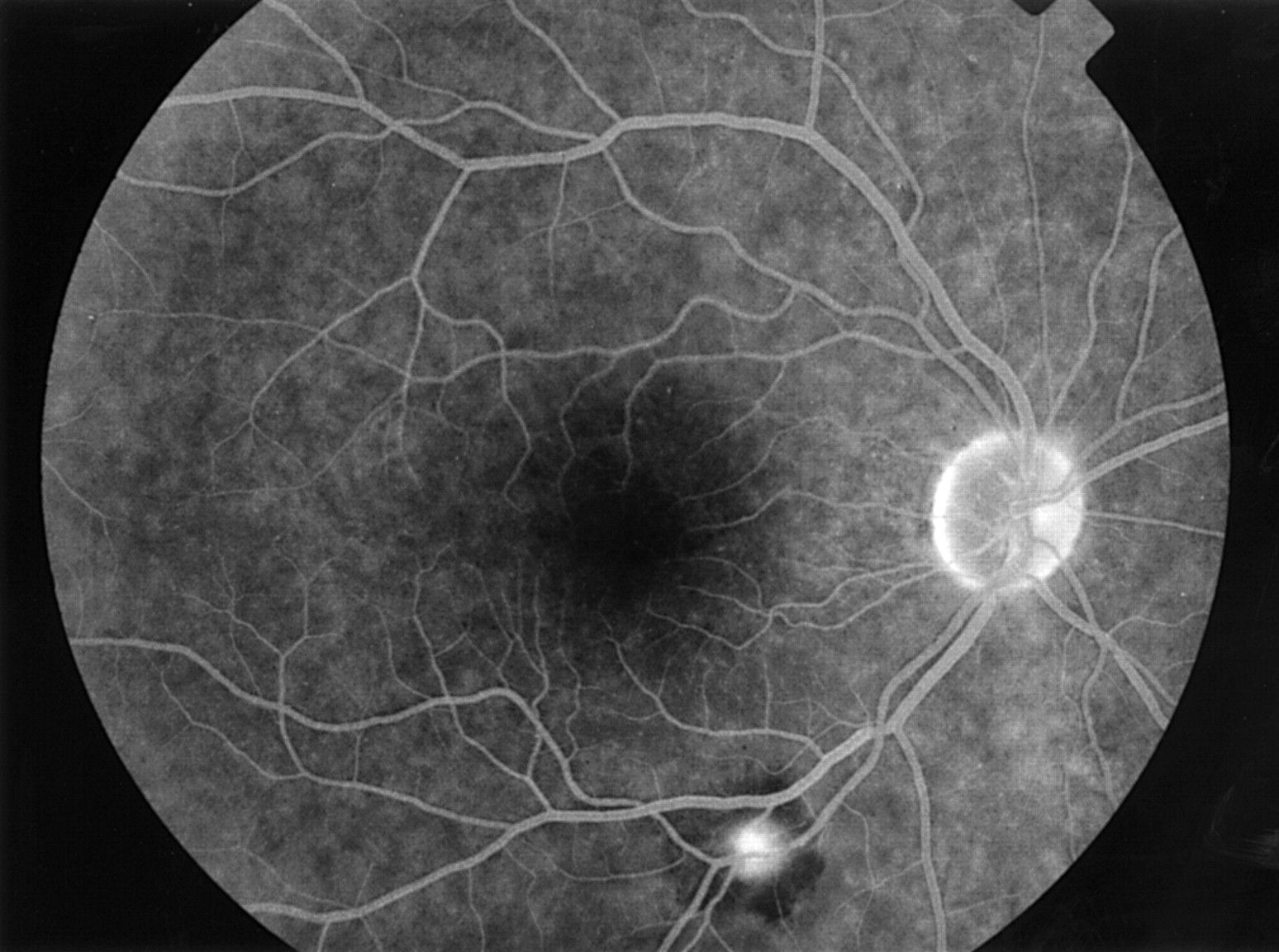
Characteristic funduscopic findings in posterior segment involvement include periphlebitis, sometimes subclinical and only visible on fluorescein angiography (Fig 2), associated with typical segmental cuffing or more extensive sheathing and perivenous exudates, which are usually indicated as “candle wax drippings” (Fig2).86-88 The term “taches de bougie” was originally used by Franceschetti in 1949, to indicate multiple small round chorioretinal lesions, which were located in the peripheral retina and were not associated with retinal vessels86; this original term was probably first translated as “candle wax drippings” and (with the original description already forgotten) subsequently used to indicate the different characteristic features of sarcoidosis—namely, perivenous exudates.

Retinal vasculitis in sarcoidosis. (A) Retinal vasculitis in a 22 year old white male patient with sarcoidosis visible as focal perivascular inflammatory infiltrates. (B) Fluorescein angiogram of this patient exhibits focal perivenous leakage in the affected areas. (C) Retinal vasculitis in a 44 year old white male patient with sarcoidosis. Late phase fluorescein angiogram. Note the multiple segmental perivascular leakage areas. (D) Sheathing of the peripheral retinal vessels surrounded by haemorrhages in a 24 year old black female patient with sarcoidosis. The retinal abnormalities normalised after periocular corticosteroid administration.
Choroidal lesions may be multifocal and located anywhere in the fundus. Round punched out lesions (the original “taches de bougies”) are frequently seen in the peripheral retina; recently the clinical entity of peripheral multifocal chorioretinitis was described in elderly white females, characterised by the presence of intraocular inflammation and multiple (>10) punched out lesions in the peripheral retina (Fig3).73 This entity was predominantly seen in elderly white female patients and was associated with the frequent occurrence of cystoid macular oedema (72%) and visual loss (42%). The association of acute posterior multifocal placoid pigment epitheliopathy (AMPPE) and sarcoidosis has also been noted.89 Optic disc leakage may be frequently observed on fluorescein angiography and might be secondary to uveitis. Differentiation from genuine optic nerve disease is required because of the therapeutical consequences. Supposedly choroidal granuloma may be observed as an elevated white mass (Fig4).90-92 The association of posterior segment and neurological involvement in sarcoidosis has been reported to be as high as 27%.62 The presentation as haemorrhagic retinopathy resembling Eals' disease has been described as well as branch and central retinal vein occlusions, development of capillary non-perfusion, and subsequent neovascularisations, which may react well to anti-inflammatory treatment (Figs 2D and 5).93-98 The development of retinal pigment detachment has also been noted. Optociliary shunts, dilated collateral veins on the optic nerve head, connecting the central retinal vein to the peripapillary choroidal venous plexus, have been reported, and may be misdiagnosed as neovascularisation of the optic disc (Fig 6).78 Arterial macroaneurysms, occurring in elderly female patients with peripheral multifocal chorioretinitis have been described, and were associated with severe cardiovascular disease (Fig 7).99 The most frequent complications of posterior segment involvement in sarcoidosis included cystoid macular oedema (76%), cataract (49%), glaucoma (36%), retinal ischaemia (16%), and neovascularisations (11%).71 Fluorescein angiography is a standard technique for monitoring the posterior segment activity; recently indocyanine green (ICG) angiographic findings in patients with posterior segment sarcoidosis demonstrated the widespread extent of choroidal involvement. ICG angiography may therefore represent an additional tool for monitoring the disease spread and activity.100

Multiple chorioretinal punched out lesions in the peripheral fundus in a 64 year old white female patient with sarcoidosis (peripheral multifocal chorioretinitis). Note the confluence of the lesions in the far periphery.

Posterior segment granulomas in sarcoidosis. (A) Peripheral choroidal nodule surrounded by several small chorioretinal lesions in a 26 year old white male patient with sarcoidosis. (B) Whitish chorioretinal nodules nasal to optic nerve head in a 55 year old white female patient with sarcoidosis. (C) Scarification of a large choroidal granuloma in 30 year old white male patient with sarcoidosis located in inferior part of the fundus and associated with secondary changes of retinal pigment epithelium.

Peripheral area of capillary non-perfusion with impeding development of the neovascular membrane in ocular sarcoidosis. Late phase fluorescein angiogram.

Optic disc and peripapillary atrophy with abnormal vessels (optociliary shunts) of the optic nerve head in a patient with juvenile onset sarcoidosis. Focal chorioretinal lesions in different stages of inflammatory activity were visible throughout the fundus and in the peripapillary area.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Arterial macroaneurysms in a patient with sarcoidosis. Late phase fluorescein angiogram.
Differential diagnosis is wide and sarcoidosis should be looked for in almost all forms of intraocular inflammation. Differentiation from other granulomatous diseases such as tuberculosis should be attempted. Patients with the clinical features of intermediate uveitis (pars planitis), multifocal chorioretinitis, Behçet's disease, syphilis, toxoplasmosis, and intraocular lymphoma should also be assessed for sarcoidosis. Birdshot chorioretinopathy may strongly resemble sarcoidosis; the true association has also been noted.101-103 Retinal abnormalities from sickle cell disease and other haemoglobinopathies in young black people may also be confused with ocular sarcoidosis.
TREATMENT OF OCULAR SARCOIDOSIS
Corticosteroids are the mainstay of therapy for ocular sarcoidosis and are given preferably in topical or periocular administration. Cycloplegics are frequently needed to prevent synechiae. The indication for systemic treatment in ocular disease includes optic neuritis and severe or topical treatment resistant posterior uveitis. The initial dosage depends on the disease severity, but often high dosage is required to induce the remission or quiescent stage of intraocular inflammation; thereafter slow tapering, and prolonged low dose treatment is usually administered to maintain the disease quiescence. For corticosteroid resistant patients, administration of low dose methotrexate therapy may be effective.55 56 There remains some debate over how to treat the patients with cystoid macular oedema; treatment will usually be initiated for visual acuity below 20/40, but possibly more early treatment is required to prevent future visual loss. The surgical intervention in ocular sarcoidosis should preferably be performed at a quiescent stage; with aggressive inflammation control before cataract surgery, IOL implantations were well tolerated and good visual results were achieved.104 The major cause of the decreased visual acuity following cataract extractions was the pre-existing glaucomatous damage and posterior segment involvement, especially the presence of cystoid macular oedema. Neovascularisation may regress with systemic anti-inflammatory treatment, though cases with retinal ischaemia, persistent neovascularisation, or vitreous haemorrhage benefit from laser treatment.105 Vitrectomy may be essential for the differentiation of sarcoidosis from intraocular malignancy, and for cases with persistent vitreous haemorrhage or resistant vitreous opacities. The effect of the removal of the vitreous on intraocular inflammation and on cystoid macular oedema has not yet been proved.
Conclusions
Ocular sarcoidosis may present with a wide variety of ocular symptoms in all parts of the eye and may be associated with chronic and progressive intraocular inflammation leading to visual deterioration. The diagnosis may be difficult owing to the absence of diagnostic criteria and the variety of presentations. Although isolated anterior uveitis is more frequently seen in black patients, the (more severe) posterior segment involvement is encountered in white patients. Visual loss is associated with posterior segment involvement and is caused by cystoid macular oedema in the majority of cases. Treatment, which has not changed during the past decades, consists mostly of symptomatic administration of corticosteroids. An evaluation of the efficacy of other therapeutic alternatives for ocular sarcoidosis is needed.