Article Text

Statistics from Altmetric.com
Diabetic retinopathy (DR) is a leading cause of blindness but it is not known why retinopathy should be so early and so severe a complication of diabetes. The sensory loss caused by minute retinal lesions is part of the problem, but the diabetic changes in the brain are different and less serious than in the retina, often described as an outpost of the brain. This has led to the concept of a local factor being responsible for the microvasculopathy of DR. There are physiological factors unique to the retina, and it is suggested below how these, by causing hypoxia very early in diabetes, could activate cytokines that produce the microvascular changes. If retinal hypoxia is an important causal factor in the production of DR, prevention of hypoxia should ameliorate DR. This hypothesis predicts that retinitis pigmentosa (RP) should prevent DR. Both old and new work is described, which indicates that this is in fact the case, thus pointing to new, simple, and effective ways of delaying the progress of diabetic retinopathy.
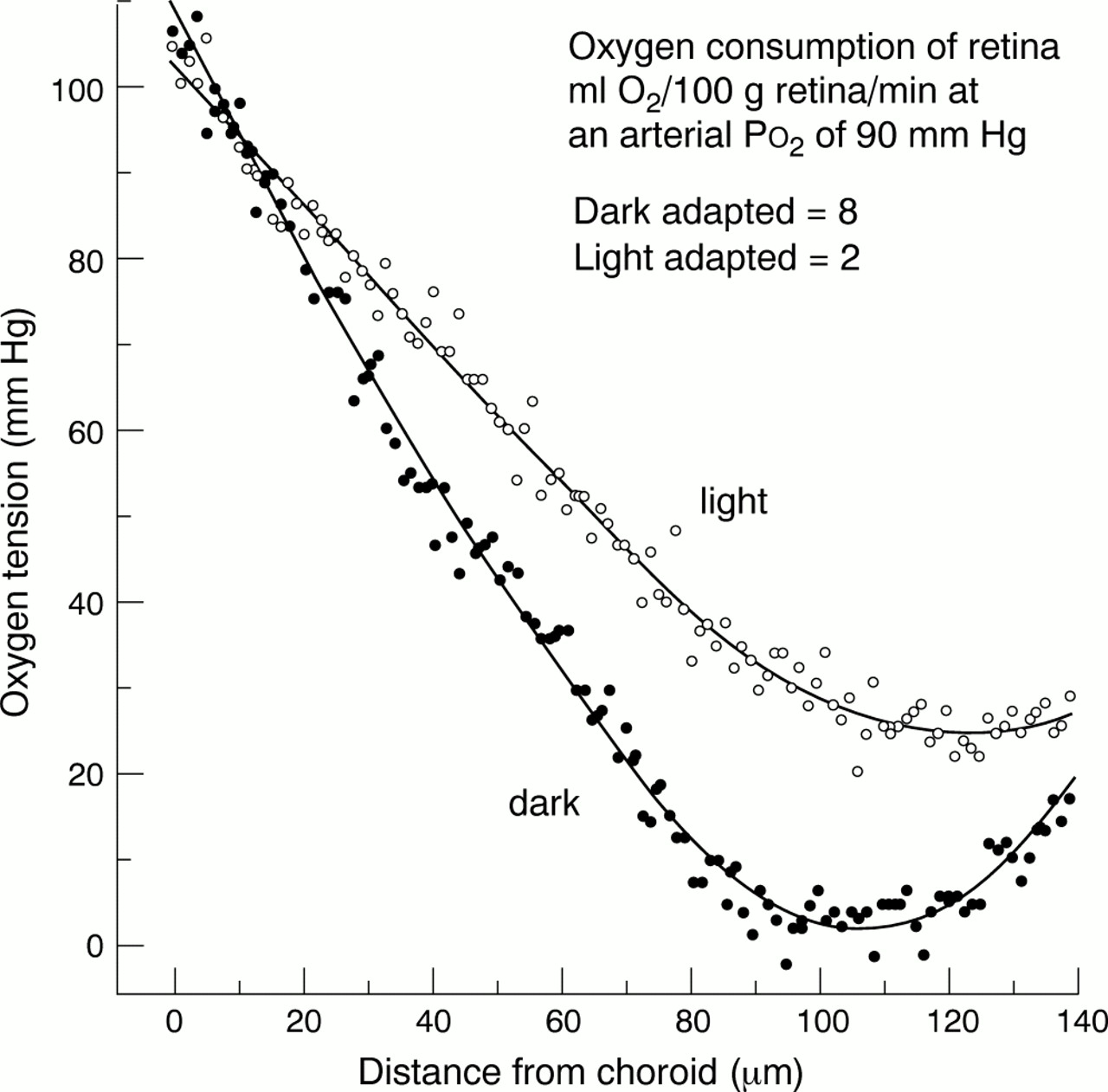
Direct comparison of retinal and brain capillaries taken from diabetics show very considerable differences1 (Table 1), which indicate a “local factor” in the development of DR. It has been suggested2 that the local factor is related to what is unique to the retina, the photoreceptors. The 120 million rods have the highest metabolic rate of any cell in the body. In darkness, the outer limb membrane “leaks”, causing an inward “dark current”. This current is reduced by light, and at normal photopic levels is shut off completely. In full dark adaptation sodium ions and water enter the outer limb at a maximal rate, and are pumped out in the inner limb.3 The entire cytosol volume is pumped in about 15 seconds.4 This process requires a great deal of energy and a large oxygen supply. However, the rods are avascular. Despite the “wall” of blood in the choroid and the extensive ramification of the central artery of the retina, oxygen tension ([Po2]) among the unusually large mitochondria of the inner limb is essentially zero.5-7When a flash of light is delivered to the retina, [Po2] abruptly rises as the pumps slow down.8 Figure 1 shows this effect.
Differences between retinal and brain capillaries of 10 diabetic and normal dogs. Duration of diabetes: 5 years
{kind=link}
Recent work9-13 confirms this result, and also shows that in dark adaptation the receptor layer removes considerable amounts of oxygen from the inner retina so [Po2] changes with illumination can also be seen there.14 The unusually low [Po2] would of course affect many other cells adversely. Although some compensatory mechanisms may occur, very small decreases in normal oxygen supply affect retinal function. Thus, dark adaptation is incomplete at reduced air pressures equivalent to heights of only 3000 feet—aeroplane cabins are pressurised to 7000 feet—and photopic vision is only affected at about 12 000 feet.215-17 Mild unilateral carotid insufficiency causes a unilateral and reversible loss of rod threshold.1819 Polycythaemia vera produces a rheological change in red blood cells that effectively slightly reduces oxygen delivery: and, again, rod threshold increases, reversibly.20
Thus, the normal retina in dark adaptation uses so much oxygen that it borders on the pathologically anoxic. There is also evidence that, in diabetics, the retina suffers from oxygen lack before the onset of clinical DR. The electroretinogram becomes abnormal years before funduscopic changes can be seen.21 Both (photopic) contrast sensitivity22 and colour vision23are impaired even before any microaneurysms are present; and inhaling oxygen from a face mask partially reverses the raised threshold, although the extra oxygen carried to the retina is very small. Scotopic threshold has been known for half a century to be more seriously affected.1624 Recent investigations suggest that in mild DR, the threshold initially falls normally, but then recovery ceases, as it would if dark adaptation was occurring in the presence of a dim background light.17 Direct measurement of [Po2] in diabetic cat retina has disclosed reductions below the normal even in areas without evident capillary dropout. Thus, it is certain that the diabetic retina rather than being hyperoxic, as is commonly supposed, suffers oxygen lack.25-27 All recent work on human retinal blood flow shows that at very early stages of diabetic retinopathy, the circulating blood volume decreases slightly.28-31 The vasodilatation that occurs slightly later (in the presence of hyperglycaemia) is quite different. The hypoxia of early diabetic retina is easily explained. The known rheological changes in red blood cells32 (similar to polycythaemia vera), the increase in the thickness of capillary basal membranes,1 the alteration in the dissociation curve for glycosylated haemoglobin, and the extra demand for oxygen consequent on higher intracellular glucose levels could all cause functional hypoxia.32-35
If the early diabetic retina is hypoxic, does this matter? Epidemiological evidence shows that anaemia is (after hyperglycaemia) the greatest risk factor for the development of DR, but the most compelling evidence comes from panretinal photocoagulation (PRP). It was thought that PRP might work by closing leaking vessels and thus preventing the release of vasoformative factors,3637 but it now appears that the major cause of the efficiency of the treatment is that it reduces the retinal oxygen need and increases retinal [Po2].38
How could a decrease in retinal [Po2] cause diabetic retinopathy? This question has recently been answered by studies on the cytokine vascular endothelial growth factor (VEGF). VEGF levels increase in diabetic retina.39-42 They are enormous in vitreous samples from patients with proliferative DR,4344 are related to disease severity45and decrease after successful treatment.46 The rise occurs in humans and animals before the development of microvascular changes.47-51 Injections of VEGF can produce proliferative retinopathy. VEGF is produced by glia (Müller) cells, which show signs of hyperactivity in early DR at the level of the outer retina, and the receptors for VEGF are located on small vessels in the inner retina.4652-54 Here VEGF damages endothelial cells and cause leakage.55 VEGF is upregulated by hyperglycaemia5156 and, very significantly, by hypoxia and the mechanism is such that brief episodes of hypoxia can cause prolonged periods of upregulation.57 Mechanisms of action of VEGF have been elucidated.58-63 Blockage of VEGF receptors can prevent the retinal neovascularisation and the increased permeability caused by VEGF.6465
Thus, a complete chain of evidence implicates VEGF in the causation of DR, and the initial reduction in oxygen supply (in relation to need) could act to initiate the changes which then enter a vicious circle. It is interesting that the nephropathy which accompanies DR is also apparently caused by VEGF acting on glomerular membranes.66 Of course this is not to deny the place of other causes of DR (which are outside the scope of this review), but the importance of real anoxia preceding DR, independent of hyperglycaemia, has not previously been highlighted.
A hypothesis which suggests that retinal anoxia is a major underlying cause of DR can be tested. Reduction in retinal metabolism should be associated with a decrease in the development of DR. This is in fact the case. Retinal scars, choroiditis, or advanced glaucoma are all epidemiologically associated with a reduction in DR. Crucially, since rod activity is supposedly responsible for DR, in the absence of rods DR should not occur. Anecdotal evidence of this nature goes back to 1966,67-69 and in diabetic people who also suffer RP or choroideraemia, the changes of DR never occur (personal communications). However, there is only one recent letter70 on this subject, and like the other citations67-69 it deals only with proliferative retinopathy. Accordingly, further work was done to better establish the absence of DR in patients with RP.
A large group of inherited degenerative retinal diseases (all called RP) are characterised by an early loss of rod function. Loss of cone vision occurs later, with scotomata, although the function of the fovea is often maintained until middle age.70-73 Although the phenotypes are similar, molecular genetic studies have shown a wide variety of basic abnormalities. Thus, more than 30% of patients with autosomal dominant inheritance have abnormalities in the gene (Rho) coding for rhodopsin.73-76 However, mutations in genes coding for other photoreceptor structural proteins and proteins concerned with phototransduction also produce RP.77-87 In addition, a number of examples have been reported in which quite different phenotypes can be caused by genetic defects that also cause clinical RP.88 RP is also associated with defects of quite different genes—for example, those concerned with the production of myosin89 or the mutations in mitochondrial DNA found in families with maternal inheritance of retinal abnormality (MIDD).90-93
RP and diabetes occur independently. Therefore, although each condition is relatively common, the number of patients with both conditions seen by doctors with large practices is quite small. To identify a relatively large number of patients, advertisements were placed in the internet “chat rooms” of RP patients, and contact made with the websites of patient support groups, requesting patients with both conditions to reply to the author. Some of these were reprinted in braille news sheets. Those replying were sent a brief questionnaire to determine if both diseases were present (Date of birth? Date of diagnosis of diabetes? Do you take insulin? What drugs do you take? At what age were you night blind? What is your vision now? Could you attend a centre to have special photographs of your eyes?). The questionnaire also asked for names and addresses of diabetologists and ophthalmologists who were caring for the patients, and requested permission to approach these people with more detailed questionnaires. The attending doctors were approached to determine the ocular and diabetic status of the patients, even when the patients also gave the details themselves and were informed and definite. The doctors were asked to confirm the type of RP, the date at which the patient became night blind or the field became constricted, the degree of diabetic control, the type of diabetes, and the presence or absence of other diabetic complications. They were asked to provide fundus photographs. The investigation conformed to the Declaration of Helsinki. Ethical approval was obtained locally and from the Foundation Fighting Blindness.
Names and addresses of over 200 patients were received, mostly from the patients' support groups. The largest number, 168, came from the Foundation Fighting Blindness (USA). Sixty seven patients replied to the initial questionnaire. They lived in a number of different countries—USA, New Zealand, Australia, Ireland, France, Germany, Switzerland, and the UK. The proportion of “take up” was lowest for the USA (20%). This is possibly because the FFB registers have been in existence longest, and patients on the register having moved house more, or have died, grown older, and been unable to respond to written communications.
Of the replies, it was established that seven did not have RP. All these were French (RP is the French acronym for proliferative DR). Of the remainder, two patients' relatives were unwilling to provide further information and some doctors, although repeatedly approached, did not respond. The final results are shown in Table 2. No patient had any DR. In view of the relatively high age of onset in the IDDM group (35 years), the identification of the type of diabetes in some patients is suspect. In some cases, only ophthalmologists replied, and some stated that they did not know if the patients had non-ocular complications of diabetes, or they did not know the details of treatment. However, copies of medical notes and fundus photographs (of varying quality) were also provided in a number of cases. No patient had had the four field standard photographs mandatory for new epidemiological studies and no patient was willing to attend a (remote) centre for such photographs to be taken. Thus the patients' present ophthalmic state is well but not perfectly documented. The reduced number of patients in the last rows of Table 2 reflect the lack of information in the doctors' files about events in their patients' past. Although various types of RP inheritance were represented—autosomal dominant, autosomal recessive, and X linked, as well as Usher's type 1—in only one case had genetic screening been performed. Two cases of Lawrence-Moon-Bartlett-Biedel syndrome are included. The mean age of the patients responding is high, and so therefore is the duration of their diabetes. Since about 40% of those responding with appropriate information had other diabetic complications, the total absence of any signs of DR is striking. There were no microaneurysms in the sample, no exudates of any type, and no haemorrhages in the retina. Patients were positive that they had never had any retinal abnormalities. Individual histories are illustrative: thus, one patient developed diabetes in early childhood, and 45 years later has no DR, although she has diabetic nephropathy requiring dialysis, and diabetic cardiopathy. Another, aged 78, who developed diabetes aged 5 has no DR. Another, aged 68, with autosomal dominant inheritance reported his night vision became poor 16 years after the onset of diabetes, confirming70 the considerable protection against the appearance of DR in patients with RP.
Details of a survey of patients with diabetes mellitus and retinitis pigmentosa
Although the sample is still small, it is double that of all the other reports combined.67-70 It is unrepresentative, if only for the age of the respondents, though for the purposes of the investigation the long duration of DM is an advantage. The results completely bear out the belief of ophthalmologists specialising in retinal degenerations that RP protects against DR and supplement the previous survey of typical RP,70 which was concerned only with the presence of proliferative retinopathy. It is reasonably certain that replies were obtained for people with a number of different mutations in different chromosomes.
By making the assumptions (as in Holmes-Walkeret al92) that previous epidemiological studies9495 of DR are appropriate for this survey (that is, conservatively, ∼75 % of patients should have fundal changes of DR 15 years after diagnosis) the probability of obtaining, by chance, a population of 55 cases with RP and no DR is extremely small. The crude binomial probability is 4/10 000 (0.7527). Type II patients may develop diabetes when the retina is relatively non-functional and atrophic. However, in type I, the mean interval between night blindness and the onset of DM is only ∼7 years and thus many patients had relatively large areas of partially functioning retina when DM began. In three cases DM developed in childhood, and these patients must have had considerable retinal function and diabetes for more than 10 years. Thus, although the methodology of a retrospective survey is not ideal, it does also highlight the point that these patients have never exhibited evidence of background retinopathy. There are so many genetic changes which cause RP that the only single unifying cause for the protection is the loss of photoreceptors, importantly rods. In many cases of autosomal dominant RP 50% of rods vanish with an elevation of rod threshold by only 0.3 log unit (due to a loss in the “quantum catch”).217 This degree of night blindness should be as effective as a PRP which “burnt” 50% of the retina, and illustrates why RP is so protective.
Very occasionally, patients with RP may develop neovascularisation at the optic disc which can regress or develop, but this phenomenon is not understood and the process quite different from DR.96-98Maternally inherited diabetes and deafness (MIDD3243), a mitochondrial disease of adult life, sometimes also causes a pigmentary retinopathy that differs considerably from the more common forms of RP in which rod loss occurs early. Thus, where electroretinograms (ERGs) have been performed on patients with MIDD, they are abnormal in only 4/13, and rod and cone ERGs are equally affected. In nine of 24 there is either reduced visual acuity or an abnormal visual cortical evoked potential, suggesting earlier macular involvement than in classic RP. Of 24 MIDD cases with DM and pigmentary retinopathy reported, in two different investigations, five have non-proliferative DR,91-93 so they are significantly different (χ2 test) from ordinary RP (Table 2). In cases where there is no RP, the incidence of DR is higher, so even incomplete rod loss is partially protective against DR. The cause of MIDD is an abnormality in the reaction centres that produce a proton gradient in the mitochondrion. The variability of the symptomatology is thought to be due to the simultaneous occurrence of normal and affected reaction centres in the same mitochondrion, and the normal/abnormal ratio in different tissues and cells. The way diabetes develops has recently been elucidated.99 The fairly frequent occurrence of lesions in photoreceptors is not unexpected in view of the intense metabolic requirements of the inner limb mitochondria.
Comparison of MIDD and the nuclear genetic disturbances may help to discriminate between rival hypotheses21770 of the formation of DR. It has been suggested that an important factor in the production of DR is the production of free radicals. The proposed sequence was that in DM increased glycolysis leads to acidosis and the retina is also hyperoxic. Under such conditions proton gradients induce free radicals,99 and these cause DR. The absence of DR in RP was explained by the suggestion that loss of photoreceptors reduced glycolysis and decreased the production of free radicals. In MIDD the basic loss is an inability to make ATP through oxidative phosphorylation and loss of the proton gradient. It is suggested that glycolysis increases.2193 If the production of free radicals depends on the establishment of a proton gradient, MIDD might be expected to be protective against DR but this is the case only when retinal degeneration occurs. Thus, the observations on MIDD make the importance of free radicals to DR less likely and are consistent with the anoxia/VEGF hypothesis described here.
In life, dark adaptation and its accompanying low [Po2] occurs mostly during sleep.217 In turn, this suggests that non-destructive methods of reducing rod dark current could help prevent DR by increasing retinal oxygen tension when it is lowest.100-102 The dark current is maintained by cyclic guanydyl monophosphate (cGMP), which is destroyed after the absorption of photons. Intracellular calcium accelerates cGMP cyclase and thus the formation of cGMP, but when the pores in the rod outer limb membrane close under the influence of light, calcium entry is reduced and a Na-Ca exchanger greatly reduces [Ca2+]in. This is one important mechanism of light adaptation.3Reducing rod guanydyl cyclase activity, or pharmacologically preventing calcium entry, or preventing full dark adaptation by a continuous low level of background light should be an effective form of decreasing peak outer limb retinal oxygen demand. Such interventions could thus slow the progress of diabetic retinopathy. The latter method is simple, inexpensive, and particularly appropriate for the developing world.
Note added at proof stage:
Since this article was submitted it has been shown103that in a group of long term diabetics who do not develop DR, the upregulation of VEGF by anoxia is largely absent, thus lending strong support to the hypothesis developed above.
Acknowledgments
The author thanks the Foundation Fighting Blindness Inc, the British Retinitis Pigmentosa Society, and Retinitis Pigmentosa International for the facilities offered, and Dr JE Wolf, Dr J Heckenlively, Dr CS Bain, Dr R Tzekov, and Mr AMP Hamilton, FRCS, for their assistance in contacting patients and for helpful discussions.