Article Text

Statistics from Altmetric.com
- ASD, anterior segment dysgenesis
- ASMD, anterior segment mesenchymal dysgenesis
- ASOD, anterior segment ocular dysgenesis
- ERG, electoretinogram
- STR, short tandem repeat
- VEP, visual evoked potential
The phrase anterior segment dysgenesis (ASD), also sometimes known as anterior segment ocular or mesenchymal dysgenesis (ASOD or ASMD, OMIM #107250), was first used in 1981 by Hittner and colleagues to describe a range of developmental defects in structures at the front of the eye.1 These defects are thought to result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to the cornea, iris, and other components of the anterior chamber during eye development.2,3 Conditions falling within the ASD spectrum include aniridia, posterior embryotoxon, Axenfeld’s anomaly, Reiger’s anomaly/syndrome, Peters’ anomaly, and iridogoniodysgenesis. Aniridia (OMIM #106210) ranges from almost complete absence of the iris, through enlargement and irregularity of the pupil mimicking a coloboma, to small slit-like defects in the anterior layer visible only with a slit lamp. In Axenfeld’s anomaly (OMIM #109120), the Schwalbe’s line is prominent and centrally displaced (posterior embryotoxon) with peripheral iris strands fused to it. Eye signs in Rieger’s anomaly (OMIM #109120) patients may include malformations of the anterior chamber angle and aqueous drainage structures (iridogoniodysgenesis), microcornea, iris hypoplasia, eccentric pupil (corectopia), iris tears (polycoria), and iridocorneal tissue adhesions traversing the anterior chamber angle.4,5 In Reiger’s syndrome (OMIM #180500), patients have ASD in association with underdeveloped or misshapen teeth (hypodontia) and may also have hearing loss, heart defects and skeletal abnormalities. Peters’ anomaly (OMIM #604229) consists of a central corneal leucoma, absence of the posterior corneal stroma and Descemet membrane, and varying degrees of iris and lenticular attachment to the posterior cornea. All of these conditions carry with them an increased risk of glaucoma owing to abnormalities in the Schlemm’s canal and trabecular meshwork.
Human ASD phenotypes are genetically heterogeneous,6 resulting from mutations in seven different transcription factor genes (PAX6, PITX2, PITX3, FOXC1, FOXE3, LXM1B, and MAF) a cytochrome enzyme gene (CYP1B1), and the human homologue of the Drosophila “eyes absent” gene (EYA1),6,7 the exact function of which is not yet known. Mutations in two further genes, Bmp4 and Rxr-alpha,8–10 cause ASD in mice, and linkage studies in families have implicated further, as yet unidentified, genes in human ASD. However, genetic analysis has also shown that the same mutation in different patients can cause different forms of ASD, suggesting that the different clinical categories of ASD represent a spectrum of severity in what is essentially the same underlying condition.11 Furthermore, specific phenotypes are not the result of mutations in only a single gene. Peters’ anomaly can result from a mutation in any one of four different genes, PAX6, PITX2, CYP1B1, or FOXC1, and each of these have also been implicated in other ASD phenotypes. One consistent finding is that, as a cause of human inherited eye disease, these conditions are almost always inherited in a dominant fashion. The only clear exception is mutations in the CYP1B1 gene, which lead to Peters’ anomaly and primary congenital glaucoma. In mice, homozygosity for mutations in these genes generally results in more severe phenotypes such as anophthalmia or microphthalmia, suggesting an additive rather than a truly dominant effect.12
Key points
-
We document a new phenotype of recessively inherited foveal hypoplasia and anterior segment dysgenesis in a large consanguineous pedigree.
-
Five members of the pedigree presented with nystagmus and poor vision. These individuals, their siblings, and their parents underwent full systemic and ophthalmic examination. Ophthalmic examination revealed foveal hypoplasia and anterior segment dysgenesis in all five cases, in the absence of any other abnormalities. Parents were unaffected.
-
Linkage analysis identified a region on chromosome 16q23.2-24.2 that showed highly significant co-segregation with the disease in this family. Multipoint linkage analysis generated a lod score of 5.51 at D16S511. Recombination events defined a 22.1 cM/6.5 Mb interval between markers D16S3098 and D16S2621, which includes the transcription factor FOXC2. However, sequencing of this single exon gene in patients‘ DNA revealed no mutations.
-
The combination of developmental abnormalities in both anterior and posterior eye structures suggests a defect in early ocular development and morphogenesis.
By contrast, foveal hypoplasia is a less well documented condition affecting the retina. It is characterised by an absent or abnormal foveomacular reflex, unclear definition of the foveomacular area, and presence of capillaries running abnormally close to the macula. Patients have decreased visual acuity and congenital nystagmus. When associated with other eye conditions such as albinism, incontinentia pigmenti, trisomy 8 mosaic syndrome, or Goldenhar-Gorlin syndrome, foveal hypoplasia is a recognised cause of poor vision in early childhood.13 It has also been documented in patients with mutations in PAX6, both in isolation,14 and in association with ASD.15 Isolated foveal hypoplasia is said to be rare and sporadic,16,17 although it has been suggested that this condition may be an underdiagnosed cause of congenital nystagmus.17
We now report a consanguineous Asian (Pakistani) family with five affected members in three separate sibships, segregating foveal hypoplasia in a clearly recessive fashion (fig 1). All of the affected individuals also have anterior segment abnormalities with variable severity. Two have Axenfeld’s anomaly, while the remaining three have posterior embryotoxon. This combination of abnormalities in structures at both the front and back of the eye has been seen before only in association with dominant mutations in PAX6, which was excluded by linkage analysis at the outset of the study. We therefore carried out further genetic analysis in this family in order to locate another gene with an important role in eye development.
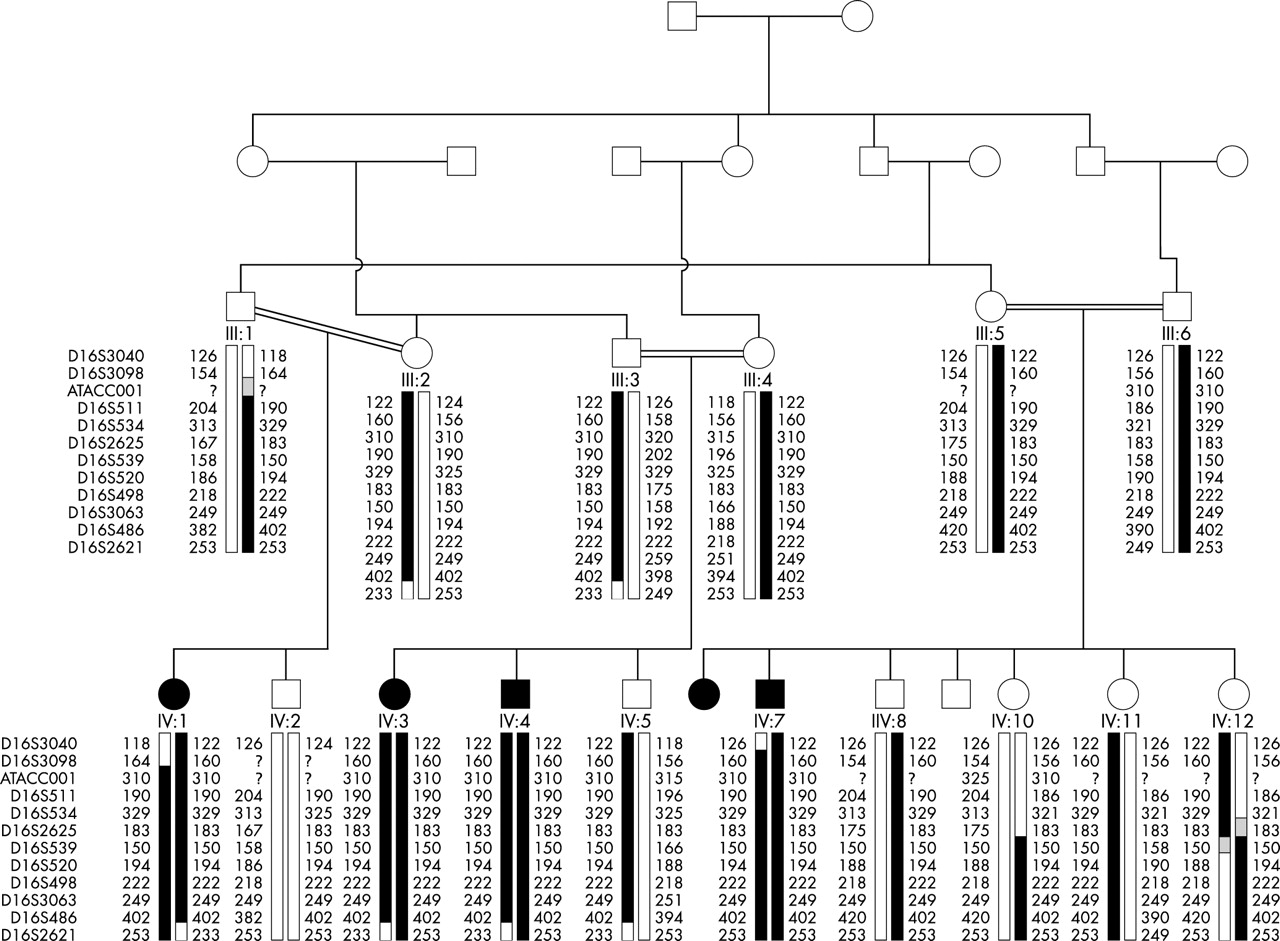
Haplotypes for markers on chromosome 16q in the family with foveal hypoplasia and anterior segment dysgenesis.
PATIENTS AND METHODS
Phenotypic assessment of the primary family
Affected members of this pedigree presented with nystagmus and poor vision secondary to foveal hypoplasia. They were subjected to full systemic and neurological paediatric examination. In addition, the four oldest family members were subjected to a full ophthalmic examination with slit lamp together with refraction. All other siblings and their parents from affected sibships were examined using a portable slit lamp. The youngest patient was examined by portable slit lamp and direct ophthalmoscopy. Fundus photographs were taken, and visual function tests, including electroretinograms (ERGs) and visual evoked potentials (VEPs) were assessed using standard protocols.18
Genotyping and linkage analysis
Blood samples were obtained from sixteen members of the pedigree, including four affected individuals, and genomic DNA was extracted from peripheral blood leukocytes by standard methods. Members of the family were then genotyped for STR markers from version 11 of the Marshfield screening set (http://www.marshfield-clinic.org/research/ genetics/). Further refinement and haplotype analysis was performed using fluorescently tagged microsatellite markers derived from the Marshfield19 and DeCODE20 human genome maps. PCR reactions were carried out in a 25 μl volume with 50 ng of DNA, 20 pmol of each primer (one fluorescent and one unlabelled primer in each pair), 200 μM of each dNTP, 10 mM Tris-HCl pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.01% gelatin, and 1 U Taq DNA polymerase (Invitrogen). After the initial denaturation step at 95°C for 3 minutes, the samples were processed through 35 cycles of 94°C for 30 seconds, 50–60°C for 30 seconds, and 72°C for 40 seconds. A final extension step was performed at 72°C for 10 minutes. Following amplification, PCR products were resolved on an ABI 377 sequencer and analysed using Genscan 2.0.2 and Genotyper 1.1.1 software (Applied Biosystems). Data was stored and haplotypes were assembled using Cyrillic 2.1 software (Cherwell Scientific). The Cyrillic package was also used to produce pedigree files for linkage analysis. Analysis of the full genome search data was carried out using the Genehunter program21 through the Genome Linkage User Environment, available via the Human Genome Mapping website (http://www.hgmp.mrc.ac.uk/). Once linkage had been established, more detailed two point and multipoint linkage mapping was carried out using the programmes MLINK and LinkMap, from the LINKAGE suit.22 Genetic distances in these analyses were taken from the Marshfield19 and DeCODE20 human genome maps. Allele frequencies were estimated by genotyping a panel of 20 unrelated Pakistani Asian individuals.
Mutation analysis
Owing to its large size and high GC content, the single exon FOXC2 gene was amplified in three overlapping segments. Reactions were carried out in a 50 μl volume with 50 ng of DNA, 20 pmol of each primer, 200 mM of each dNTP, 10 mM Tris-HCl pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.01% gelatin, 6% DMSO, 1 M betaine and 1 U Taq DNA polymerase (Invitrogen). After the initial denaturation step at 95°C for 3 minutes, the samples were processed through 35 cycles of 94°C for 30 seconds and 62°C for 2 minutes. A final extension step was performed at 68°C for 10 minutes. Both the forward and reverse strands of the PCR products were sequenced using a DYEnamicTM ET dye terminator kit (MegaBACE, Amersham Biosciences) and sequences were resolved on a MegaBace 500 sequencer (Amersham Pharmacia Biotech). Sequences were read on both strands independently by two researchers and compared with normal control.
RESULTS
Clinical findings
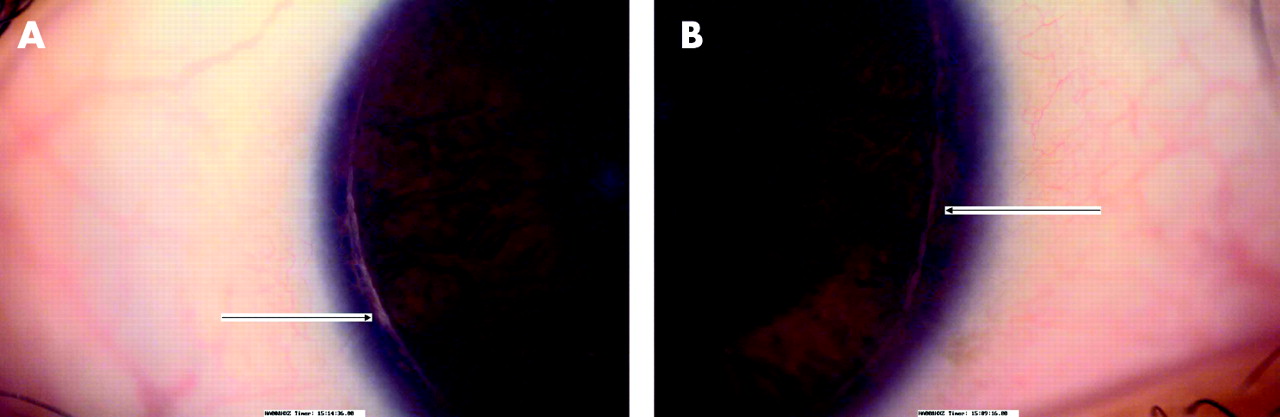
All five affected members of the family described here have a history of poor vision and nystagmus since birth. A full systemic and neurological examination was normal in each case. With refraction, the best corrected visual acuity in the four patients examined ranges from 20/120 in patient IV-1 to 20/400 in patient IV-7. All of these patients have refractive problems. Patient IV-3 has high hypermetropia while patients IV-1, IV-4, and IV-7 have astigmatism. All the affected individuals have anterior segment dysgenesis. Patients IV-3, IV-4, and IV-6 have posterior embryotoxon (fig 2A) while patient’s IV-1 and IV-7 have Axenfeld’s anomaly (fig 2B). The posterior embryotoxon (thickened Schwalbe’s line) is more pronounced in patients with Axenfeld’s anomaly. Intraocular pressures are normal in all patients and there is no evidence of developmental glaucoma as yet. In the fundus, the discs appear normal, but there is foveal hypoplasia with a poor or absent foveal reflex, unclear distinction of the foveomacular area and a poorly defined foveal avascular zone (fig 3A–D). The clinical findings are summarised in table 1.
Primers used to amplify the FOXC2 coding sequence
Clinical findings in affected individuals

(A) Thickened Schwalbe’s line (arrowed) indicative of posterior embryotoxon in patient IV-4. (B) Axenfeld’s anomaly in patient IV-1, with peripheral iris strands attaching to the prominent Schwalbe’s line (posterior embryotoxon).
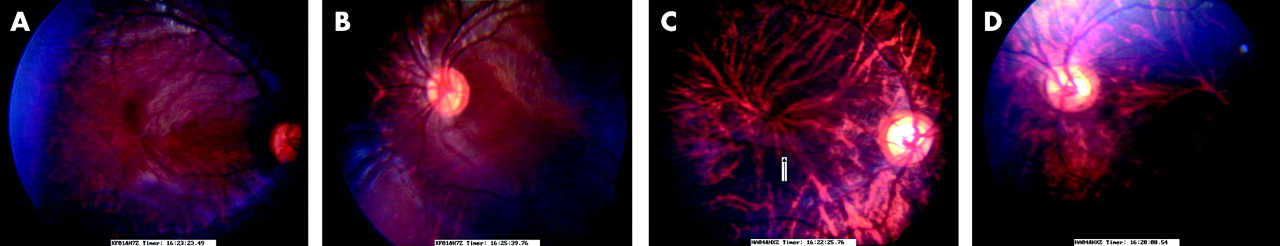
(A,B) Fundus photographs of foveal hypoplasia showing an abnormal macula with poor foveal reflex in patient IV-1. (C,D) Fundus photographs of patient IV-4, showing poor foveal reflex and a vessel (arrowed) crossing the macular area, indicative of foveal hypoplasia.
As a test of visual function, ERGs and VEPs were carried out in the four older affected members and three parents. The parents showed no measurable abnormality in these tests. All affected individuals have decreased ERG amplitudes and poor VEP amplitudes with increased latency, with some variation in severity.
Linkage analysis
The combination of foveal hypoplasia and ASD type abnormalities initially suggested the involvement of the PAX6 gene, where a similar combination of symptoms had been documented, albeit with dominant inheritance. We therefore tested markers immediately adjacent to this and two other candidate ASD genes, PITX2 and FOXC1. Multipoint analysis of these data conclusively excluded these genes from involvement.22
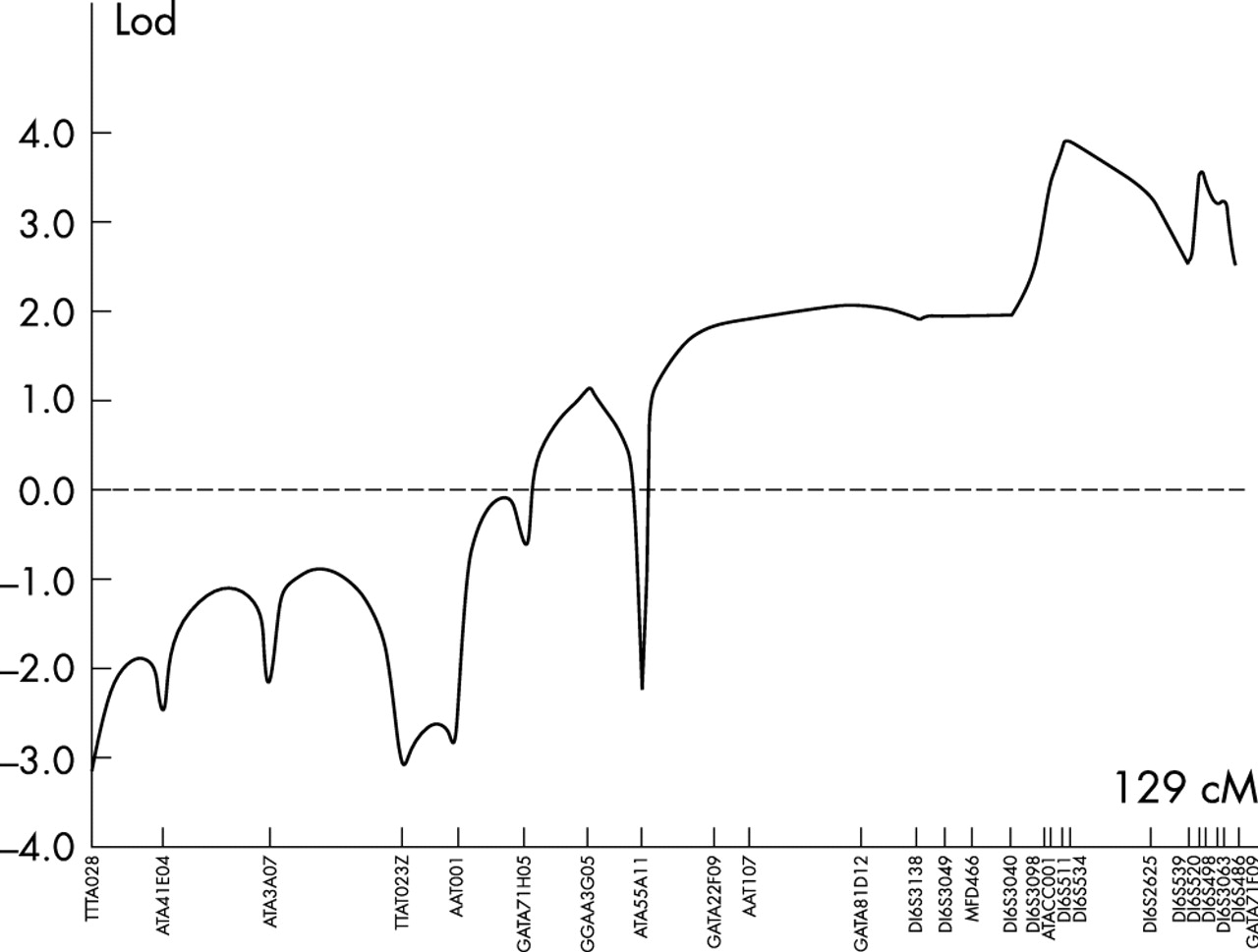
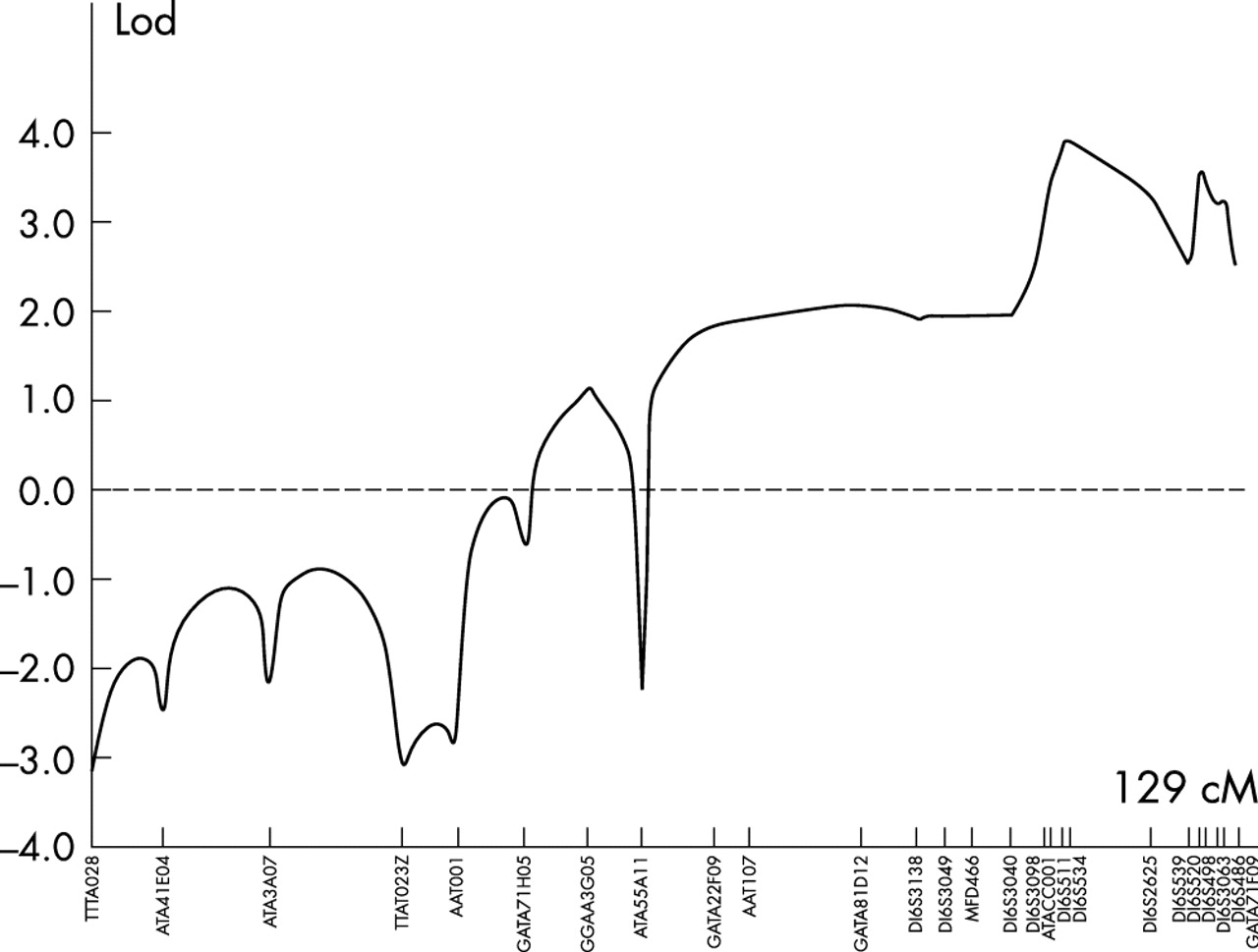
In order to locate the defective gene, a whole genome linkage search was therefore carried out. All 16 members of the pedigree for whom DNA was available were genotyped for the polymorphic short tandem repeat (STR) markers in version 11 of the Marshfield screening set (http://www.marshfield-clinic.org/research/genetics). This set constitutes 407 polymorphic STRs spanning the genome with an average spacing of 9 cM. Statistically significant exclusion (lod score ⩽2) was obtained across most of the genome, with no significant positive lod scores except those confined to a region towards the telomere of chromosome 16q. Analysis of data from the initial genomewide search revealed an interval of 30 cM between markers MFD466 (100 cM from 16pter) and marker 16S2621 (130 cM from 16pter) in which multipoint linkage analysis gave a lod score of 2.5. Genoyping additional markers refined the proximal end of the interval to marker D16S3098 (108.3 cM), reducing the region to 22.11 cM (108.3–130.41 cM from 16pter). Haplotypes for 16q markers in the family are shown in fig 3.
To carry out linkage analysis with all chromosome 16 markers, the family was reduced to three subpedigrees and data were analysed using the Genehunter program. This analysis shown in fig 4 generated a maximum lod score of 4.01 at the marker D16S511. Using the two point lod score programme MLINK, scores of >3 were obtained with markers D1S3098, D1S511, D1S534, D1S520, and D1S486, in the region, with a maximum lod score of 4.36 with marker D16S511. Multipoint analysis with markers D16S511, D16S534, and D16S520 using LinkMap gave a maximum lod score of 5.51 at D16S511, proving beyond reasonable doubt the existence of a new locus for foveal hypoplasia and anterior segment dysgenesis in this family.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lod scores for markers spanning chromosome 16 generated using the linkage program Genehunter. Genetic distances were derived from the Marshfield genetic map.
Candidate gene screening
The forkhead family of transcription factors share a conserved 100 amino acid domain with unique DNA binding characteristics, known as the FKH box (hence the acronym FOX). The first forkhead gene was identified in Drosophila by Weigel and colleagues.23 Subsequently many of these genes have been implicated in various developmental disorders in humans and other species.24–26 As detailed above, two FOX genes have been implicated in ASD disorders. FOXC1 mutations have been found in patients with Reiger’s syndrome, Axenfeld’s and Peters’ anomaly, iridogoniodysgenesis, and iris hypoplasia with glaucoma. In addition, a single FOXE3 mutation was identified in a family with posterior embryotoxon and cataracts.26
Another member of this gene group, FOXC2, maps to human chromosome 16q24.1, within the critical interval defined by genetic analysis in this family. Nonsense mutations in FOXC2 cause lymphoedema-distichiasis syndrome, a dominant disorder that presents with lymphoedema of the limbs, a double row of eyelashes, and a number of other complications, including ptosis and photophobia.27–29 In additon, FOXC2 mutations have been known to cause alterations in human corneal thickness.30 However, mice heterozygous for a targeted deletion of foxc2 (+/−) have anterior segment abnormalities similar to those reported in humans with Axenfeld-Rieger anomaly.31 These observations made FOXC2 a strong candidate for involvement in the phenotype described here. We therefore screened the entire coding sequence of this single exon, 501 codon gene for mutations in patients. However, no mutations were found.
DISCUSSION
Here we describe a large consanguineous pedigree segregating foveal hypoplasia and ASD in a recessive fashion. Albinism, the commonest cause of foveal hypoplasia, was excluded by clinical examination and VEP. This combination of recessively inherited isolated foveal hypoplasia, in the absence of other known syndromic features and in association with ASD, represents a new clinical entity. To date, nine genes have been implicated in ASD-type conditions, as described above, but of these only the transcription factor PAX6, when mutated, can cause a similar combination of defects in the anterior and posterior segments of the eye. The dominant Gly64Val mutation in PAX6 causes foveal hypoplasia, cataracts, and corneal epithelial changes comparable to those seen in aniridia.32 By inference, it therefore seems likely that the defective protein in this family, like PAX6, will have a significant role in the developmental processes that control the formation of the eye.
Foveal hypoplasia is found in association with albinism and a number of syndromic human conditions. Of particular note, Gillespie syndrome is a combination of aniridia, foveal hypoplasia, cerebellar ataxia, and mental deficiency.33 Patients in the family described here showed no neuronal deficit on examination, distinguishing this phenotype from Gillespie syndrome. Nevertheless, it remains possible that these two conditions are in fact allelic, as no locus has yet been mapped for Gillespie syndrome. In addition, two patient series, comprising 24 patients with isolated foveal hypoplasia, have been reported.16,17 The mode of inheritance and any potential environmental contribution in these cases remain to be resolved, although the lack of family history is consistent with recessive inheritance. The authors of both papers suggest that foveal hypoplasia may be an underdiagnosed cause of congenital nystagmus. In a third patient series with a similar phenotype, De Pool and colleagues described what they term an ateliotic macula, a developmental disorder involving absence of the foveal reflex, thinning of the retina, and poor vision at birth. Interestingly, of their case series of seven patients, two are siblings and are the children of first cousins, implying recessive inheritance. In addition, two other unrelated cases in the same series have an ateliotic macula in association with ASD.34
Distal 16q is already a chromosomal region of interest to those working on ASD. The first authors to make this link, Ferguson and Hicks, described in 1987 a patient with a partial trisomy of 16q associated with Rieger’s syndrome.35 Later, deletion of 16q23.1–16q24.2 was found in a patient with bilateral coloboma of the iris, short stature, moderate developmental delay, and minor craniofacial anomalies.36 Linkage to markers at 16q24 has been seen in a family with autosomal dominant Rieger’s syndrome, though no lod scores were shown and this abstracted result has not as yet been published in full.37 Whether these conditions and the one we now report are allelic remains to be determined. One gene located on 16q has already been implicated in ASD. Mutations in the transcription factor MAF, at 16q23.1, have been found to cause cataracts, iris coloboma, and anterior segment dysgenesis.38 This gene initially appeared to be a promising candidate for involvement in the phenotype described above, but was found to lie 1.5 Mb/4.5 cM proximal to the new locus.
FOXC2 was considered a logical candidate for involvement in the phenotype described, based on its position, its homology to FOXC1, and the ASD phenotype seen in heterozygous mice with a targeted deletion of FOXC2. Furthermore, anterior segment ocular anomalies have been reported in patients with lymphoedema−distichiasis syndrome caused by mutations in FOXC2.30 These were milder than those caused by FOXC1 mutations and were not associated with glaucoma, similar to the findings in heterozygous knockout mice.31 We found no coding sequence changes in affected individuals, but mutations upstream or downstream of the gene, which alter expression, cannot be ruled out. It may be pertinent that all ASD mutations in transcription factors documented to date have caused dominant phenotypes, so the recessive mode of inheritance in this family may imply a different disease mechanism.
In summary, we have identified a locus on chromosome 16q23.2–24.2 at which a new phenotype of recessively inherited foveal hypoplasia and anterior segment dysgenesis cosegregate. Mutations in the coding sequence of the gene encoding transcription factor FOXC2 have been excluded. At least 37 positional candidate genes lie within this locus, making further genetic studies in this and other families with similar conditions a priority. The identification of the gene involved may contribute to a better understanding of eye embryogenesis and development.
Acknowledgments
We thank the Marshfield Genotyping service for conducting the initial genome linkage search in this family.
REFERENCES
Footnotes
-
↵* Present address, Department of Anatomy and Cell Biology, University of Bergen, Norway
-
This study was supported by Wellcome Trust Grants 071590 and 073477.
-
Conflicts of interest: none declared
-
The first two authors contributed equally to this work.